CATALYSTS FOR OLEFIN POLYMERIZATION FIELD OF THE DISCLOSURE The present disclosure relates to new indenyl ligands, bisindenyl ligands comprising said indenyl ligands, complexes thereof, and catalysts comprising those complexes. The present disclosure also relates to the use of the new bisindenyl metallocene catalysts for the production of polypropylene homopolymers or propylene copolymers, especially with ethylene, in particular heterophasic polypropylene, with high activity levels, high molecular weight, and hence low MFR, and with ideal melting points. The catalysts are especially useful in the manufacture of polypropylene homopolymer of high melting point. BACKGROUND OF THE DISCLOSURE Metallocene catalysts have been used to manufacture polyolefins for many years. Countless academic and patent publications describe the use of these catalysts in olefin polymerization. Metallocenes are now used industrially and polyethylenes and polypropylenes in particular are often produced using cyclopentadienyl based catalyst systems with different substitution patterns. WO2007116034 describes C2-symmetric complexes bearing 5-methoxy substituents and 6-tert-butyl substituents. Such catalysts produce relatively low melting hPP of 148 to 150 °C. WO2018091684 describes C2-symmetric rac-Me2Si(2-Me-4-(3,5-Me2Ph)-5-OMe-6-tBu- Ind)2ZrCl2 complex, that produces hPP with low Tm of 150-151 °C. WO2001048034 describes C1-symmetric bisindenyl complexes having 1-methyl and 1- isopropyl as 2-substituents. Although the produced hPP can have Tm as high as 160 °C, the catalyst activity is always very low. WO2005058916 describes a series of C1-symmetric bisindenyl complexes combining one 2-iso-propyl-4-aryl-indenyl ligand and one 2-methyl-4-aryl-indacenyl ligand. While Tm of hPP is somewhat high varying from 152 to 160°C, catalyst activities are invariably low. WO0202576 describes the C
2-symmetric rac-Me
2Si(2-iPr-4-(3,5-Me
2Ph)-Ind)
2ZrCl
2. This complex provides a catalyst of very low activity and hPP of relatively low T
m of 152 to 154°C). The teaching from this patent is that iPr groups lower catalyst activity without increasing hPP T
m. EP1421090 describes C
2-symmetric rac-Me
2Si(2-iPr-4-(2-R-Ph)-Ind)
2ZrCl
2. These complexes provide catalysts of very low activity although hPP T
m is relatively high (157- 159°C).
US9745390B2, US9464145B2, US9249239B2, EP3022235B1, and EP3022238B1 all describe bridged, C
1-symmetric bisindenyl complexes having two different 2-substituents, but none of the complexes contain a methoxy group. EP3572441 is also describing C
1-symmetric bisindenyl complexes having two different 2- substituents being one methyl and one isopropyl containing a Et
2Si bridge. T
m of hPP is relatively low at 155 to 156 °C. While these prior art catalysts have their advantages, they lack in isoselectivity, generating hPP with a relatively low melting and crystallisation temperature due to the formation of insertion regiodefects in the PP chains. The present inventors sought new metallocenes, which provide high isoselectivity without compromising productivity, especially in the case of the homopolymerization of propylene or in the case of copolymerization between propylene and ethylene. The desired catalysts should also have improved performance in the production of high melting temperature and high molecular weight polypropylene homopolymers (hPP). The desired catalysts should also have improved performance in the production of propylene-ethylene copolymers, for instance having high activity for high Mw copolymer products. The desired catalysts should also provide propylene-ethylene copolymers having high molecular weight. Further, the desired catalysts should also be able to produce hPP with Tm at least 157 °C. BRIEF DESCRIPTION OF THE DISCLOSURE An object of the present disclosure is to provide new ligands, metallocene complexes, and hence catalysts to overcome the above problems. The object of the disclosure is achieved by metallocene complexes of formula (I), polymerisation catalyst comprising said metallocene complex of formula (I), and process for polymerisation of polypropylene optionally with comonomers which are characterized by what is stated in the independent claims. The preferred embodiments of the disclosure are disclosed in the dependent claims. It was surprisingly found that specific modification of either C2-symmetric or C1-symmetric, preferably C2-symmetric, metallocenes incorporating alpha-branched alkyl substituent on one of the 2-positions of the ligand, in combination with specific substitution of the 4, 5 and 6 ligand positions, provide the desired properties. The identified metallocene complexes when included in a polymerization catalyst, preferably supported catalyst system, composed of the said specific class of metallocene complexes in combination with an aluminium containing cocatalyst have improved
polymerization behavior, higher catalyst productivity, improved performance in the production of propylene homopolymers, propylene random copolymers and heterophasic propylene copolymers compared to systems known in the art, enabling the production of propylene-ethylene copolymers of high Mw, thus being ideal for the production of propylene random copolymers, especially propylene-ethylene random copolymers, and also suitably heterophasic propylene copolymers. The specific catalyst system gives a higher flexibility/freedom in the design of propylene polymers than prior art catalyst systems. An advantage of the disclosure is that these metallocenes allow the production of propylene polymers having high isotacticity, in particular upon MAO activation, producing homopolymer polypropylene (hPP) with higher T
m. BRIEF DESCRIPTION OF THE DRAWINGS In the following the disclosure will be described in greater detail by means of preferred embodiments with reference to the accompanying drawings, in which Figure 1 shows melting point of hPP as a function of MFR2; Figure 2 shows metallocene polymerisation activity versus hPP MFR2. DEFINITIONS Throughout the description, the following definitions are employed: The term “C1-20-hydrocarbyl” includes C1-C20-alkyl, C2-C20-alkenyl, C2-C20-alkynyl, C3-C20- cycloalkyl, C3-C20-cycloalkenyl, C6-C20-aryl, C7-C20-alkylaryl, and C7-C20-arylalkyl groups or of course mixtures of these groups such as cycloalkyl substituted by alkyl. Unless otherwise stated, preferred C1-C20-hydrocarbyl groups are C1-C20-alkyl, C4-C20-cycloalkyl, C5-C20-cycloalkyl-alkyl groups, C7-C20-alkylaryl groups, C7-C20-arylalkyl groups, and C6-20- aryl groups, especially C1-C10-alkyl groups, C6-C10-aryl groups, and C7-C12-arylalkyl groups, e.g. C1-C8-alkyl groups. Most especially preferred hydrocarbyl groups are methyl, ethyl, propyl, isopropyl, tert-butyl, isobutyl, C5-C6-cycloalkyl, cyclohexylmethyl, phenyl, and benzyl. The term “C1-C10-hydrocarbyl” includes C1-C10-alkyl, C2-C10-alkenyl, C2-C10-alkynyl, C3-C10- cycloalkyl, C3-C10-cycloalkenyl, C6-C10-aryl, C7-C10-alkylaryl, and C7-C10-arylalkyl groups or of course mixtures of these groups such as cycloalkyl substituted by alkyl. Unless otherwise stated, preferred C1-C10-hydrocarbyl groups are C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl groups, C7-C10-alkylaryl groups, C7-C10-arylalkyl groups, and C6-C10- aryl groups, especially C1-C6-alkyl groups, C6-aryl groups, and C7-C10-arylalkyl groups, e.g.
C
1-C
6-alkyl groups. Most especially preferred hydrocarbyl groups are methyl, ethyl, propyl, isopropyl, tert-butyl, isobutyl, C
5-C
6-cycloalkyl, cyclohexylmethyl, phenyl, and benzyl. It is to be noted that linear and branched hydrocarbyl groups cannot contain cyclic units. Aliphatic hydrocarbyl groups cannot contain aryl rings. The term “heteroatoms of Group 14-16 of the Periodic Table” includes for example Si, N, O or S. The term “C
4-C
8 ring” as used herein in connection to -R1 2Si-, refers to cyclic groups containing 4 to 8 carbon atoms and a Si atom and includes for example silacycloalkanediyls, such as silacyclobutane, silacyclopentane, or 9-silafluorene. The term “halogen” includes fluoro, chloro, bromo, and iodo groups, especially chloro or fluoro groups, when relating to the complex definition. The oxidation state of the metal ion is governed primarily by the nature of the metal ion in question and the stability of the individual oxidation states of each metal ion. It is appreciated that in the complexes of the invention, the metal ion is coordinated by ligands X to satisfy the valence of the metal ion and to fill its available coordination sites. The nature of these sigma-ligands can vary greatly. The numbering of these rings will be evident from the structures indicated herein. Catalyst activity is defined in this application to be the amount of polymer produced/g catalyst/h. Catalyst metal activity is defined here to be the amount of polymer produced/g Metal/h. The term productivity is also known to be used to indicate the catalyst activity although herein it designates the amount of polymer produced per unit weight of catalyst. The term “molecular weight” is used herein to refer to weight average molecular weight Mw unless otherwise stated. DETAILED DESCRIPTION OF THE DISCLOSURE This invention relates to a series of new ligands, metallocene complexes, and hence catalysts that are ideal for the polymerization of propylene. Metallocene catalyst complexes The complexes of the invention can be asymmetrical or symmetrical. Asymmetrical means simply that the two indenyl ligands forming the metallocene are different, that is, each indenyl ligand bears a set of substituents that are either chemically different, or located in different positions with respect to the other indenyl ligand. Symmetrical complexes are based on two identical indenyl ligands.
The metallocene complexes of the invention are preferably chiral, racemic bridged bisindenyl C
1-symmetric metallocenes in their anti-configuration. Although the complexes of the invention are formally C
1-symmetric, the complexes ideally retain a pseudo-C
2- symmetry since they maintain C
2-symmetry in close proximity of the metal center although not at the ligand periphery. By nature of their chemistry both anti and syn enantiomer pairs (in case of C
1-symmetric complexes) are formed during the synthesis of the complexes. For the purpose of this invention, racemic-anti means that the two indenyl ligands are oriented in opposite directions with respect to the cyclopentadienyl-metal-cyclopentadienyl plane, while racemic-syn means that the two indenyl ligands are oriented in the same direction with respect to the cyclopentadienyl-metal-cyclopentadienyl plane, as shown in the scheme below.
Racemic Anti Racemic Syn Preferred metallocene catalyst complexes are in the anti-configuration. The metallocene complexes of the invention are preferably employed as the racemic-anti- isomers. Ideally, therefore at least 95 mol%, such as at least 98 mol%, especially at least 99 mol% of the metallocene catalyst complex is in the racemic anti-isomeric form. For the purpose of this invention, the numbering scheme of the indenyl ligands is the following:
The present metallocene catalyst complexes require the combination of two distinctive features of the ligand framework: 1: specific 5, 6 substitution of the indenyl ligands, and 2: one alpha-branched substituent on the 2-position of one of the indenyl ligands. The present invention accordingly relates to metallocene complexes of formula (I)
wherein Mt is Zr or Hf, preferably Zr; X is a sigma ligand R
1 are each independently, same or different from each other, C
1-C
20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C
4-C
8-ring; R
2 is linear C1-C20-, preferably C1-C10-hydrocarbyl;
R
2’ is alpha-branched C
3-C
10-hydrocarbyl or SiH(R
21’)
2, with R
21’ being each independently, same or different from each other, C
1-C
10-hydrocarbyl; n are each independently an integer from 1 to 5; R
3 and R
4 are each independently, same or different from each other, H, C
1-C
10- hydrocarbyl, -OR
31, -SR
31, or –N(R
31)
2, with R
31 being C
1-C
10-hydrocarbyl, whereby at least one R
3 and at least one R
4 is not hydrogen; L is O or S; R
51 and R
51’ are each independently, same of different from each other, C
1-C
10- hydrocarbyl; and R
6 and R
6’ are each independently, same or different from each other, C(R
61)
3 or -OR
61, with R
61 being each independently, same of different from each other, linear or branched C
1-C
6-alkyl; or adjacent LR
51 and R
6 and/or LR
51’ and R
6’ form together a -O[C(R
62)
2]
mO- group, with R62 being each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R
1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R
1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R
1 groups are the same. Most preferably, both R
1 are methyl.
Preferably R
2 is CH
2-R
21, with R
21 being H, linear C
1-C
6-alkyl, such as methyl, ethyl, n- propyl, i-propyl, n-butyl, preferably R
21 being H or linear C
1-C
3-alkyl; more preferably R
2 is methyl or ethyl, most preferably methyl. Preferably R
2’ is CH(R
21’)
2 or SiH(R
21’)
2, with R
21’ being each independently, same of different from each other, linear or branched C
1-C
6-alkyl, C
3-C
8-cycloalkyl, or C
6-C
9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R
21’ being linear or branched C
1-C
6-alkyl; more preferably, R
2’ is CH(CH
3)
2 or SiHMe
2, even more preferably CH(CH
3)
2. It is preferred that each n is independently, same or different from each other, an integer from 1 to 3, such as 1, 2 or 3. Preferably, the R
3 and R
4 substituents of the respective phenyl ring are in the 3´-, 4´-, and/or 5´-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the two phenyl rings have different substitution patterns. It is therefore preferred when n is 1, the only R
3 and/or R
4 group, respectively, is preferably in the para position. If n is 2, then the two R
3 and/or R
4 groups, respectively are preferably in the meta positions. Preferably R
3 and R
4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C10-aryl, or -OR
31, with R
31 being C1-C4-hydrocarbyl; even more preferably, each R
3 and R
4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R
3 and at least one R
4 is not H. As noted above, R
51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R
51 and R
51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R
51 and R
51’ are the same. Most preferably, both R
51 and R
51’ are methyl.
Preferably R
6 and R
6’ are each independently, same or different from each other, C(R
61)
3 or –OR
61, with R
61being each independently, same of different from each other, linear C
1- C
3-alkyl; more preferably methyl. It is preferred that R
6 and R
6’ are the same. Advantageously, both R
6 and R
6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R
62)
2]
mO- group, each R
62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-a)
wherein Mt is Zr or Hf; X is a sigma ligand R
1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R
2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R
2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R
21’)2, with R
21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R
3 and R
4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR
31, -SR
31, or –N(R
31)
2, with R
31 being C
1-C
10-hydrocarbyl, whereby at least one R
3 and at least one R
4 is not H; R
51 and R
51’ are each independently, same of different from each other, C
1-C
10- hydrocarbyl; and
R
6 and R
6’ are each independently, same or different from each other, C(R
61)
3 or -OR
61, with R
61 being each independently, same of different from each other, linear or branched C
1-C
6-alkyl; or adjacent OR
51 and R
6 and/or OR
51’ and R
6’ form together a –O[C(R
62)
2]
mO- group, with R62 being each independently, same or different from each other, H or linear or branched C
1- C
6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I-a), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-a) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C
1-C
6-alkoxy, or R´ group, where R´ is C
1-C
6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R
1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R
1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R
1 groups are the same. Most preferably, both R
1 are methyl. Preferably R
2 is CH2-R
21, with R
21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R
21 being H or linear C1-C3-alkyl; more preferably R
2 is methyl or ethyl, most preferably methyl. Preferably R
2’ is CH(R
21’)2 or SiH(R
21’)2, with R
21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R
21’ being linear or branched C1-C6-alkyl; more preferably, R
2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R
3 and R
4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR
31, with R
31 being C1-C4-hydrocarbyl; even more preferably, each R
3 and R
4 are each independently, same or different from each
other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R
3 and at least one R
4 is not H. Preferably, the R
3 and R
4 substituents of the respective phenyl ring are in the 3´-, 4´-, and/or 5´-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. It is therefore preferred if one or two R
3 and/or R
4 groups is H. If two R
3 and/or R
4 groups are H then the remaining R
3 and/or R
4 group, respectively, is preferably in the para position. If one R
3 and/or R
4 group is H then the remaining R
3 and/or R
4 groups are preferably in the meta positions. Advantageously one or two R
3 on the phenyl group are not H, when two R
3 are not H preferably these two R
3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the indenyl moiety preferably one or two R
4 on the phenyl group are not H, more preferably two R
4 are not H, and most preferably these two R
4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. As mentioned above, R
51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R
51 and R
51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R
51 and R
51’ are the same. Most preferably, both R
51 and R
51’ are methyl. Preferably R
6 and R
6’ are each independently, same or different from each other, C(R
61)3 or –OR
61, with R
61 being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R
6 and R
6’ are the same. Advantageously, both R
6 and R
6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R
62)2]mO- substituent, each R
62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2.
Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-b)
(I-b) wherein Mt is Zr or Hf; X is a sigma ligand R
1 are each independently, same or different from each other, C
1-C
20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C
4-C
8-ring; R
3 and R
4 are each independently H, C
1-C
10-hydrocarbyl, -OR
31, -SR
31, or –N(R
31)
2, with R
31 being C
1-C
10-hydrocarbyl, whereby at least one R
3 and at least one R
4 is not hydrogen; R
51 and R
51’ are each independently, same of different from each other, C
1-C
10- hydrocarbyl; and R
6 and R
6’ are each independently, same or different from each other, C(R
61)
3 or -OR
61, with R
61 being each independently, same of different from each other, linear or branched C
1-C
6-alkyl; or adjacent OR
51 and R
6 and/or OR
51’ and R
6’ form together a -O[C(R
62)2]mO- group, wherein R
62 are each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I-b), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-b) it is preferred if Mt is Zr or Hf, preferably Zr.
Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C
1-C
6-alkoxy, or R´ group, where R´ is C
1-C
6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R
1 are each independently, same or different from each other, C
1-C
10- hydrocarbyl, more preferably C
1-C
10-alkyl, C
4-C
10-cycloalkyl, C
5-C
10-cycloalkyl-alkyl, C
7- C
10-arylalkyl, C
6-C
10-aryl, or C
7-C
10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C
3-C
8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C
1-C
6-alkyl, C
5-C
6-cycloalkyl, or C
6-aryl. In an embodiment each R
1 is independently, same or different from each other, C
1-C
10-alkyl, optionally substituted with C
1-C
10-alkoxy. It is preferred that both R
1 groups are the same. Most preferably, both R
1 are methyl. Preferably R
3 and R
4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR
31, with R
31 being C1-C4-hydrocarbyl; more preferably H, linear or branched C1-C4-alkyl, or -OR
31, with R
31 being C1-C4-hydrocarbyl; even more preferably, each R
3 and R
4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R
3 and at least one R
4 is not H. Preferably, the R
3 and R
4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. It is therefore preferred if one or two R
3 and/or R
4 groups is H. If two R
3 and/or R
4 groups are H then the remaining R
3 and/or R
4 group, respectively, is preferably in the para position. If one R
3 and/or R
4 group is H then the remaining R
3 and/or R
4 groups are preferably in the meta positions. Advantageously one or two R
3 on the phenyl group are not H, more preferably R
3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl.
For the indenyl moiety preferably one or two R
4 on the phenyl group are not H, more preferably two R
4 are not H, and most preferably these two R
4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl
. As mentioned above, R
51’ is C
1-C
10-hydrocarbyl, for example, a linear or branched C
1-C
10- hydrocarbyl. Preferably R
51 and R
51’ are each independently, same of different from each other, linear or branched C
1-C
6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C
7-C
10-arylalkyl, C
7-C
10-alkylaryl, or C
6-C
10-aryl, more preferably linear C
1-C
6-alkyl, branched C
3-C
6-alkyl, or C
6-aryl, even more preferably linear C
1-C
4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R
51 and R
51’ are the same. Most preferably, both R
51 and R
51’ are methyl. Preferably R
6 and R
6’ are each independently, same or different from each other, C(R
61)
3 or –OR
61, with R
61 being each independently, same of different from each other, linear C
1- C
3-alkyl; more preferably methyl. It is preferred that R
6 and R
6’ are the same. Advantageously, both R
6 and R
6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R
62)2]mO- substituent, each R
62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (l-c)
(I-c) wherein Mt is Zr or Hf; X is a sigma ligand
R
1 are each independently, same or different from each other, C
1-C
20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C
4-C
8-ring; R
2 is linear C
1-C
20-, preferably C
1-C
10-hydrocarbyl; R
2’ is alpha-branched C
1-C
10-hydrocarbyl or SiH(R
21’)
2, with R
21’ being each independently, same or different from each other, C
1-C
10-hydrocarbyl; R
3 and R
4 are each independently, same or different from each other, C
1-C
10-hydrocarbyl group, -OR
31, -SR
31, or -N(R
31)
2, with R
31 being C
1-C
10-hydrocarbyl; R
51 and R
51’ are each independently, same of different from each other, C
1-C
10- hydrocarbyl; and R
6 and R
6’ are each independently, same or different from each other, C(R
61)
3, with R61 being each independently, same of different from each other, linear or branched C
1-C
6- alkyl. For the above-defined metallocene complexes of formula (I-c), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-c) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R
1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R
1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R
1 groups are the same. Most preferably, both R
1 are methyl. Preferably R
2 is CH2-R
21, with R
21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R
21 being H or linear C1-C3-alkyl; more preferably R
2 is methyl or ethyl, most preferably methyl.
Preferably R
2’ is CH(R
21’)
2 or SiH(R
21’)
2, wherein R
21’ are each independently, same of different from each other, linear or branched C
1-C
6-alkyl, C
3-C
8-cycloalkyl, or C
6-C
9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R
21’ being linear or branched C
1-C
6-alkyl; more preferably, R
2’ is CH(CH
3)
2 or SiHMe
2, even more preferably CH(CH
3)
2. Preferably R
3 and R
4 are each independently, same or different from each other, linear or branched C
1-C
6-alkyl, C
6-C
20-aryl, or -OR
31, with R
31 being C
1-C
4-hydrocarbyl; more preferably linear or branched C
1-C
4-alkyl or -OR
31, with R
31 being C
1-C
4-hydrocarbyl; even more preferably, each R
3 and R
4 are each independently, same or different from each other, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially methyl, or tert-butyl. Preferably, the R
3 and R
4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.3´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously one or two R
3 on the phenyl group are not H, more preferably R
3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the second indenyl moiety preferably R
4 are the same like 3´,5´-di-methyl or 3´,5´-di- tert-butyl. As mentioned above, R
51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R
51 and R
51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R
51 and R
51’ are the same. Most preferably, both R
51 and R
51’ are methyl. Preferably R
6 and R
6’ are each independently, same or different from each other, C(R
61)3, wherein R
61 are each independently, same of different from each other, linear C1-C3-alkyl; more preferably methyl. It is preferred that R
6 and R
6’ are the same. Advantageously, both R
6 and R
6’ are tert-butyl.
Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-d)
wherein Mt is Zr or Hf; X is a sigma ligand R
1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R
2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R
2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R
21’)2, with R
21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R
3 and R
4 are each independently, same or different from each other, C1-C10-hydrocarbyl group, -OR
31, -SR
31, or –N(R
31)2, with R
31 being C1-C10-hydrocarbyl. For the above-defined metallocene complexes of formula (I-d), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-d) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C
1-C
6-alkoxy, or R´ group, where R´ is C
1-C
6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl.
Preferably R
1 are each independently, same or different from each other, C
1-C
10- hydrocarbyl, more preferably C
1-C
10-alkyl, C
4-C
10-cycloalkyl, C
5-C
10-cycloalkyl-alkyl, C
7- C
10-arylalkyl, C
6-C
10-aryl, or C
7-C
10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C
3-C
8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C
1-C
6-alkyl, C
5-C
6-cycloalkyl, or C
6-aryl. In an embodiment each R
1 is independently, same or different from each other, C
1-C
10-alkyl, optionally substituted with C
1-C
10-alkoxy. It is preferred that both R
1 groups are the same. Most preferably, both R
1 are methyl. Preferably R
2 is CH
2-R
21, with R
21 being H, linear C
1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R
21 being H or linear C
1-C
3-alkyl; more preferably R
2 is methyl or ethyl, most preferably methyl. Preferably R
2’ is CH(R
21’)
2 or SiH(R
21’)
2, with R
21’ being each independently, same of different from each other, linear or branched C
1-C
6-alkyl, C
3-C
8-cycloalkyl, or C
6-C
9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R
21’ being linear or branched C1-C6-alkyl; more preferably, R
2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R
3 and R
4 are each independently, same or different from each other, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR
31, with R
31 being C1-C4-hydrocarbyl; more preferably linear or branched C1-C4-alkyl; even more preferably, each R
3 and R
4 are each independently, same or different from each other, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially methyl, or tert-butyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously R
3 are the same, like 3´,5´-di-methyl. For the indenyl moiety preferably two R
4 of each phenyl ring are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-e)
wherein R
1 are each independently, same or different from each other, C
1-C
20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form, together with the Si atom they are attached to, a C4-C8-ring; R
2 is linear C1-C20- preferably C1-C10-hydrocarbyl; R
2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R
21’)2, with R
21’ being C1-C10-hydrocarbyl; R
3 and R
4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR
31, -SR
31, or –N(R
31)
2, with R
31 being C
1-C
10-hydrocarbyl, whereby at least one R
3 and at least one R
4 is not H. For the above-defined metallocene complexes of formula (I-e), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-e) R
1 are preferably each independently, same or different from each other, C1-C10-hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10- cycloalkyl-alkyl, C7-C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert-butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R
1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R
1 groups are the same. Most preferably, both R
1 are methyl. Preferably R
2 is CH2-R
21, with R
21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R
21 being H or linear C1-C3-alkyl; more preferably R
2 is methyl or ethyl, most preferably methyl.
Preferably R
2’ is CH(R
21’)
2 or SiH(R
21’)
2, with R
21’ being each independently, same of different from each other, linear or branched C
1-C
6-alkyl, C
3-C
8-cycloalkyl, or C
6-C
9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R
21’ being linear or branched C
1-C
6-alkyl; more preferably, R
2’ is CH(CH
3)
2 or SiHMe
2, even more preferably CH(CH
3)
2. Preferably R
3 and R
4 are each independently, same or different from each other, H, linear or branched C
1-C
6-alkyl, C
6-C
20-aryl, or -OR
31, with R
31 being C
1-C
4-hydrocarbyl; more preferably H, linear or branched C
1-C
4-alkyl, or -OR
31, with R
31 being C
1-C
4-hydrocarbyl; even more preferably, each R
3 and R
4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R
3 and at least one R
4 is not H. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously R
3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the indenyl moiety preferably two R
4 of each phenyl ring are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. For the avoidance of doubt, any narrower definition of a substituent offered above can be combined with any other broad or narrowed definition of any other substituent. Throughout the disclosure above, where a narrower definition of a substituent is presented, that narrower definition is deemed disclosed in conjunction with all broader and narrower definitions of other substituents in the application. Intermediates Whilst the invention primarily relates to catalysts complexes, it will be appreciated that the ligands used to form those complexes are also new. The novel indenes of the present invention bear the combination of the distinctive features of the metallocene ligand framework: 1: 5-alkoxy indene, preferably methoxy indene, with 6-tertiary hydrocarbyl, preferably tertiary alkyl, substituent, and 2: an alpha-branched alkyl substituent on the 2-position of the indene.
The present invention accordingly further relates to indenes of formula (II)
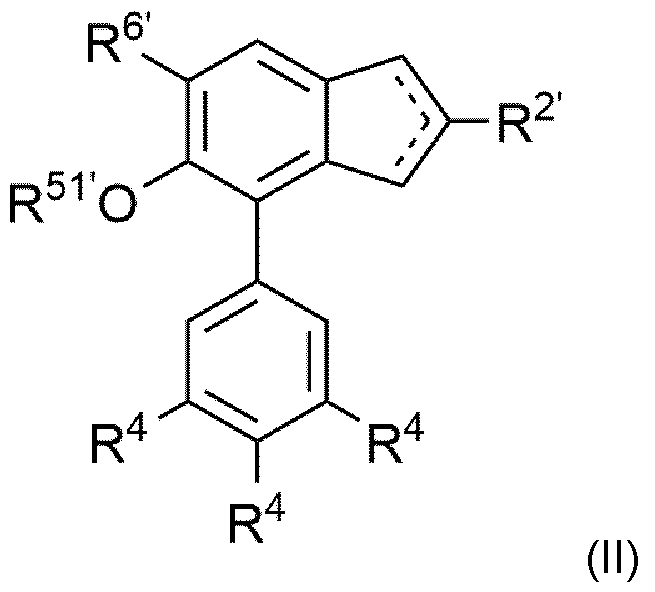
wherein the dotted lines represent a double bond present in between carbons 1 and 2 or 2 and 3 of the indenyl ring; R
2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R
21’)2, with R
21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R
4 are each independently H, C1-C10-hydrocarbyl, -OR
31, -SR
31 or –N(R
31)2 , with R
31 being C1-C10-hydrocarbyl, whereby at least one R
4 is not H; R
51’ is C1-C10-hydrocarbyl; and R
6’ is C(R
61)3, with R
61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR
5’ and R
6’ form together a –O[C(R
62)2]mO- group, with R
62 being each independently, same or different from each other, H or linear or branched C1-C6-alkyl, with m being 1 to 3. For the above-defined indenes of formula (II), the following represent preferable embodiments, which can be selected alone or in combination: Preferably R
2’ is CH(R
21’)2 or SiH(R
21’)2, with R
21’ being each independently, same or different from each other, linear or branched C1-C6-alkyl, C3-C8 cycloalkyl, or C6-C10-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R
21’ being linear or branched C
1-C
6-alkyl; more preferably, R
2’ is CH(CH
3)
2 or SiHMe
2, even more preferably CH(CH
3)
2. Preferably R
4 are each independently, same or different from each other, H, linear or branched C
1-C
6-alkyl, C
7-C
20-arylalkyl, C
7-C
20-alkylaryl, C
6-C
20-aryl, or -OR
31, being R
31 being C
1-C
10-hydrocarbyl
, whereby at least one R
3 and at least one R
4 is not H; more preferably R
4 are each independently, same or different from each other, H, linear or
branched C
1-C
6-alkyl, or C
6-C
20-aryl, even more preferably H, linear or branched C
1-C
4- alkyl, or -OR
31, with R
31 being C
1-C
4-hydrocarbyl, yet more preferably, R
4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially hydrogen, methyl, or tert-butyl, whereby at least one R
4 is not H. Preferably, the R
4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5- position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. It is therefore preferred if one or two R
4 groups is H. If two R
4 groups are H then the remaining R
4 group, respectively, is preferably in the para position. If one R
4 group is H then the remaining R
4 groups are preferably in the meta positions. For the indenyl moiety preferably one or two R
4 on the phenyl group are not H, more preferably two R
4 are not H, and most preferably these two R
4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. As mentioned above, R
51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R
51’ is linear or branched C1-C6-alkyl, such as methyl, ethyl, n- propyl, i-propyl, n-butyl, i-butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. Most preferably, R
51’ is methyl. Preferably R
6’ is C(R
61)3, with R
61 being each independently, same of different from each other, linear C1-C3-alkyl; more preferably methyl. Advantageously, R
6’ is tert-butyl. Advantageously in the -O[C(R
62)2]mO- substituent, each R
62 is independently H or methyl, preferably H, with m being1 to 3, such as 1, 2 or 3, preferably 2. Synthesis The ligands required to form the complexes and hence catalysts of the invention can be synthesized by any process and the skilled organic chemist would be able to devise various synthetic protocols for the manufacture of the necessary ligand materials. For example, WO 2007/116034 discloses the necessary chemistry. Synthetic protocols can also generally be found in WO2002/02576, WO2011/135004, WO2012/084961, WO2012/001052, WO2011/076780, WO2015/158790, WO2018/122134,
WO2019/179959, and WO2012/058740. The examples section also provides the skilled person with sufficient direction. Polymerization catalyst Viewed from a further aspect the invention provides a polymerization catalyst comprising (i) a metallocene complex of formula (I), e.g., selected from complexes of formula (I-a), (I- b), (I-c), (I-d) and (I-e); (ii) a cocatalyst comprising a group 13 element; and (iii) optionally a support. Catalyst Manufacture The metallocene complex as described above is used in combination with a suitable cocatalyst combination as described below. The metallocene catalysts can be used in supported or unsupported form. The particulate support material used is preferably an organic or inorganic material, such as silica, alumina or zirconia or a mixed oxide such as silica-alumina, in particular silica, alumina or silica- alumina. The use of a silica support is preferred. The skilled person is aware of the procedures required to support a metallocene catalyst. Especially preferably the support is a porous material so that the complex may be loaded into the pores of the support, e.g. using a process analogous to those described in WO94/14856 (Mobil), WO95/12622 (Borealis) and WO2006/097497. The particle size is not critical but is preferably in the range 5 to 200 μm, more preferably 20 to 80 μm. The use of these supports is routine in the art. Especially preferred procedures for producing such supported catalysts are those described in EP1828266, WO 2020/239598 and WO 2020/239603. Alternatively, no support is used at all. Such a catalyst can be prepared in solution, for example in an aromatic solvent like toluene, by contacting the metallocene (as a solid or as a solution) with the cocatalyst, for example methylaluminoxane or a borane or a borate salt previously dissolved in an aromatic solvent, or can be prepared by sequentially adding the dissolved catalyst components to the polymerization medium. In one aspect, no external carrier is used but the catalyst is still presented in solid particulate form. Thus, no external support material, such as inert organic or inorganic carrier, for example silica as described above is employed, but the solid catalyst is
prepared using an emulsion-solidification method. Such catalysts can be prepared as described for example in WO 2003/051934, WO 2014/060540 and WO 2019/179959. The catalyst system of the invention is preferably used in supported form. The particulate support material used is an inorganic porous support such as a silica, alumina or a mixed oxide such as silica-alumina, in particular silica. The use of a silica support is preferred. The complex may be loaded into the pores of the particulate support, e.g. using a process analogous to those described in W094/14856, W095/12622, W02006/097497 and EP18282666. The average particle size of the support such as silica support can be typically from 10 to 100 µm. However, it has turned out that special advantages can be obtained, if the support has an average particle size from 15 to 80 µm, preferably from 18 to 50 µm. The average pore size of the inorganic porous support such as silica support can be in the range from 10 to 100 nm and the pore volume from 1 to 3 mL/g. The pore diameter of the inorganic porous support such as silica support can be in the range from 20 to 40 nm. The surface area of the inorganic porous support such as silica support can be typically in the range from 100 to 400 m
2/g. Examples of suitable support materials are, for instance, ES757 produced and marketed by PQ Corporation, Sylopol 948 produced and marketed by Grace or SUNSPERA DM-L- 303 silica produced by AGC Si-Tech Co. Supports can be optionally calcined prior to the use in catalyst preparation in order to reach optimal silanol group content. The use of these supports is routine in the art. The catalyst can contain from 5 to 500 µmol, such as 10 to 100 µmol of transition metal of the metallocene per gram of support such as silica, and 3 to 15 mmol of Al per gram of support such as silica. A metallocene catalyst containing such metallocenes may be produced by a process including the steps of P1-a) combining the porous inorganic support with a first portion of the aluminoxane cocatalyst in a hydrocarbon solvent to obtain aluminoxane cocatalyst treated support, optionally followed by thermal treatment of the aluminoxane treated support; P1-b) dissolving the metallocene complex in a hydrocarbon solvent, optionally adding a second portion of the aluminoxane cocatalyst in the hydrocarbon solvent optionally the
boron containing cocatalyst wherein the amount of the first portion of the aluminoxane cocatalyst added in step a) is 75.0 to 100.0 wt% of the total amount of aluminoxane cocatalyst and the amount the second portion of the aluminoxane cocatalyst added in step b) is 0.0 to 25.0 wt% of the total amount of aluminoxane cocatalyst and the boron containing cocatalyst, when present, is added in an amount that a boron/M molar ratio of feed amounts in the range of 0.1 :1 to 10:1 is reached;; P1-c) adding the solution obtained in step b) to the aluminoxane cocatalyst treated support obtained in step a) and optionally P1-d) drying the so obtained supported catalyst system. In step P1-b) of the process, the components can be mixed in any order. The optional boron containing cocatalyst can be mixed with the metallocene complex dissolved in the hydrocarbon solvent and followed by addition the optional aluminoxane, or the metallocene complex dissolved in the hydrocarbon solvent can be mixed with the optional aluminoxane and a hydrocarbon followed by addition of boron containing cocatalyst and so on. In some embodiments, all components might be combined simultaneously. Only one impregnation step is used, i.e. the treated support of step a) is loaded only in one step with the metallocene. In a preferred aspect of the present invention the process comprises P2-a) combining the porous inorganic support with aluminoxane cocatalyst in a hydrocarbon solvent to obtain aluminoxane cocatalyst treated support, optionally followed by thermal treatment of the aluminoxane treated support, filtering off the hydrocarbon solvent, optionally washing with an aromatic solvent, repeating the filtration and washing steps to remove unreacted aluminium compounds; drying the final aluminoxane cocatalyst treated support; P2-b) dissolving the metallocene in a hydrocarbon solvent optionally adding a methylaluminoxane cocatalyst in a hydrocarbon solvent, wherein the amount of methylaluminoxane cocatalyst added in step a) is 75.0 to 100.0 wt% of the total amount of methylaluminoxane cocatalyst and the amount of aluminoxane cocatalyst added in step b) is 0.0 to 25.0 wt% of the total amount of methylaluminoxane cocatalyst, to obtain a metallocene solution optionally comprising aluminoxane cocatalyst; P2-c adding the metallocene solution to the aluminoxane cocatalyst treated support obtained in step a) and optionally P2-d) drying the so obtained supported catalyst system.
If desired, the obtained supported catalyst system may be provided as an oil slurry with a desired solid content. The solid catalyst content in the slurry may be e.g. up to 30 wt%, like up to 25 wt%. The amounts of support, aluminoxane, preferably MAO, boron containing cocatalyst and metallocene depend on the desired herein defined ratios (boron/M, Al/M, Al/SiO
2, M/SiO
2). Cocatalyst To form an active catalytic species it is normally necessary to employ a cocatalyst as is well known in the art. Cocatalysts comprising one or more compounds of Group 13 metals, like organoaluminium, organoboron, and/or borate compounds used to activate metallocene catalysts are suitable for use in this invention. Preferably, only cocatalysts comprising aluminium, like organoaluminium compounds used to activate metallocene catalysts, are utilized in this invention. According to the present invention a cocatalyst system comprising a boron containing cocatalyst and/or an aluminoxane cocatalyst is advantageously used in combination with the above defined metallocene catalyst complex. In an preferred aspect of the present invention a cocatalyst system comprising an aluminoxane cocatalyst is advantageously used in combination with the above defined metallocene catalyst complex. Thus, preferably no further cocatalysts comprising one or more compounds of Group 13 metals other than aluminium, like organoboron and/or borate compounds used to activate metallocene catalysts are comprised in the polymerization catalyst. Suitable amounts of cocatalyst will be well known to the person skilled in the art. Preferably, the amount of cocatalyst is chosen to reach below defined molar ratios. The molar ratio of Al from the aluminoxane to the metal ion (Mt) (preferably zirconium) of the metallocene Al/Mt may be in the range 10:1 to 2000:1 mol/mol, preferably 50:1 to 1000:1, and more preferably 100:1 to 600:1 mol/mol. When a boron cocatalyst is used, the molar ratio of boron (B) to the metal ion (Mt) (preferably zirconium) of the metallocene B/Mt may be in the range 0.1:1 to 10:1 mol/mol, preferably 0.3:1 to 7:1, especially 0.5:1 to 3:1 mol/mol. Even more preferably, the molar ratio of feed amounts of boron (B) to metal ion (Mt), preferably zirconium, of the metallocene B/Mt is from 0.5:1 to 2:1
Alumoxane cocatalyst The aluminoxane cocatalyst can be one of formula (A):

where n is usually from 6 to 20 and R has the meaning below. Aluminoxanes are formed on partial hydrolysis of organoaluminum compounds, for example those of the formula AlR3, AlR2Y and Al2R3Y3 where R can be, for example, C1- C10-alkyl, preferably C1-C5-alkyl, or C3-C10-cycloalkyl, C7-C12-arylalkyl or -alkylaryl and/or phenyl or naphthyl, and where Y can be hydrogen, halogen, preferably chlorine or bromine, or C1-C10-alkoxy, preferably methoxy or ethoxy. The resulting oxygen-containing aluminoxanes are not in general pure compounds but mixtures of oligomers of the formula (A). The preferred aluminoxane is methylaluminoxane (MAO). Since the aluminoxanes used according to the invention as cocatalysts are not, owing to their mode of preparation, pure compounds, the molarity of aluminoxane solutions hereinafter is based on their aluminium content. Boron containing cocatalyst According to the present invention, the aluminoxane cocatalyst can be used in combination with a boron containing cocatalyst. It will be appreciated by the person skilled in the art that where boron based cocatalysts are employed, it is normal to pre-alkylate the complex by reaction thereof with an aluminium alkyl compound, such as TIBA. This procedure is well known and any suitable aluminium alkyl, e.g. Al(C
1-C
6-alkyl)
3 can be used. Preferred aluminium alkyl compounds are triethylaluminium, tri-isobutylaluminium, tri-isohexylaluminium, tri-n-octylaluminium and tri-isooctylaluminium. Alternatively, when a borate cocatalyst is used, the metallocene complex is in its alkylated version, that is for example a dimethyl or dibenzyl metallocene complex can be used. Boron containing cocatalysts of interest include those of formula (B) BY
3 (B)
wherein Y is the same or different and is a hydrogen atom, C
1-10-haloalkyl, or C
6-C
20- haloaryl, or fluorine, chlorine, bromine or iodine. Preferred examples for Y are fluorine, trifluoromethyl, unsaturated groups such as haloaryl like p-fluorophenyl, 3,5-difluorophenyl, pentachlorophenyl, pentafluorophenyl, 3,4,5-trifluorophenyl and 3,5-di(trifluoromethyl)phenyl. Most preferably, Y are fluorine, trifluoromethyl, aromatic fluorinated groups such as p- fluorophenyl, 3,5-difluorophenyl, pentafluorophenyl, 3,4,5-trifluorophenyl and 3,5- di(trifluoromethyl)phenyl. Preferred boron containing cocatalysts of formula (B) are trifluoroborane, tris(4- fluorophenyl)borane, tris(3,5-difluorophenyl)borane, tris(2,4,6-trifluorophenyl)borane, tris(penta-fluorophenyl)borane, and/or tris(3,4,5-trifluorophenyl)borane. Particular preference is given to tris(pentafluorophenyl)borane. However it is preferred that borates are used, i.e. compounds containing a borate anion. These compounds have formula (C): Z4B
–-W
+ (C) wherein Z is a substituted phenyl derivative, said substituent being halo-C1-C6-alkyl or halogen; and W
+ is a cationic counterion. Preferably the substituents of Z are are fluoro or trifluoromethyl. Most preferably, the phenyl group is perfluorinated. The borate anion Z4B
– is preferably a weakly-coordinating anion such as tetrakis(pentafluorophenyl)borate. Suitable cationic counterions W
+ are triarylcarbenium such as triphenylcarbenium or protonated amine or aniline derivatives such as methylammonium, anilinium, dimethylammonium, diethylammonium, N- methylanilinium, diphenylammonium, N,N-dimethylanilinium, trimethylammonium, triethylammonium, tri-n- butylammonium, methyldiphenylammonium, pyridinium, p-bromo-N,N- dimethylanilinium or p-nitro-N,N-dimethylanilinium. Preferred ionic compounds which can be used according to the present invention include: tributylammoniumtetra(pentafluorophenyl)borate, tributylammoniumtetra(trifluoromethylphenyl)borate, tributylammoniumtetra(4-fluorophenyl)borate, N,N-dimethylcyclohexylammoniumtetrakis(pentafluorophenyl)borate,
N,N-dimethylbenzylammoniumtetrakis(pentafluorophenyl)borate, N,N-dimethylaniliniumtetrakis(pentafluorophenyl)borate, N,N-di(propyl)ammoniumtetrakis(pentafluorophenyl)borate, di(cyclohexyl)ammoniumtetrakist(pentafluorophenyl)borate, triphenylcarbeniumtetrakis(pentafluorophenyl)borate, or ferroceniumtetrakis(pentafluorophenyl)borate. Preference is given to triphenylcarbeniumtetrakis(pentafluorophenyl) borate, N,N- dimethylcyclohexylammoniumtetrakis(pentafluorophenyl)borate or N,N- dimethylbenzylammoniumtetrakis(pentafluorophenyl)borate. Mostly preferred are triphenylcarbeniumtetrakis(pentafluorophenyl) borate, N,N-dimethylaniliniumtetrakis(pentafluorophenyl)borate, N,N- dimethylcyclohexylammoniumtetrakis(pentafluorophenyl)borate, or N,N- dimethylbenzylammoniumtetrakis(pentafluorophenyl)borate. Polymerization The catalysts according to the invention are suitable for the production of propylene homopolymers, propylene-ethylene copolymers or propylene C4-C10 alpha olefin copolymers. Thus, the process comprises polymerizing propylene, propylene and ethylene or propylene and a C4-C10 alpha olefin. The ethylene content in such a propylene-ethylene polymer may vary depending on the desired properties of the polymer. Typically, ethylene content will range from 0.1 to 10 mol%. Especially, the catalysts of the present invention are used to manufacture propylene homopolymers or propylene copolymers with ethylene as comonomer and propylene copolymers with butene as a comonomer. Accordingly, the present disclosure relates in a further aspect to a process for producing a propylene homopolymer, a propylene random copolymer or a heterophasic propylene copolymer using the specific catalyst system, as defined before. Polymerization in the method of the invention may be effected in one or more, e.g.1, 2, or 3, polymerization reactors, using conventional polymerization techniques, e.g. gas phase, solution phase, slurry or bulk polymerization, or combinations thereof, like a combination of a slurry and at least one gas phase reactor. The process may also involve an in-line pre-polymerization step. This pre-polymerization step is a conventional step used routinely in polyolefin production plants and can be carried
out in a continuously stirred tank reactor (CSTR) or a loop reactor, from which the prepolymerized catalyst is then transferred together with the liquid monomer(s) into the main loop reactor. Prepolymerisation can be carried out at temperatures between -10 °C and 50 °C, preferably between 10 °C and 40 °C. In case of propylene polymerization in slurry reactors, like a liquid loop reactor, the reaction temperature will generally be in the range 60 to 110°C (e.g. 60 to 90°C), the reactor pressure will generally be in the range 5 to 80 bar-g (e.g.20 to 60 bar-g), and the residence time will generally be in the range 0.3 to 5 hours (e.g. 0.5 to 2 hours). The liquefied monomer is usually used as reaction medium. It is a particular feature of the invention that polymerization takes place at temperatures of at least 60 °C. For gas phase reactors, the reaction temperature used will generally be in the range 60 to 115°C (e.g.70 to 110°C), the reactor pressure will generally be in the range 10 to 30 bar- g (e.g.15 to 25 bar-g), and the residence time will generally be 0,5 to 8 hours (e.g.0,5 to 4 hours). The gas used will be the monomer optionally as mixture with a non-reactive gas such as nitrogen or propane. In addition to actual polymerization steps and reactors, the process can contain any additional polymerization steps, like a pre-polymerization step, and any further after reactor handling steps as known in the art. For solution polymerization, an aliphatic or aromatic solvent can be used to dissolve the monomer and the polymer, and the polymerization temperature will generally be in the range 80 to 200°C (e.g.90 to 150°C) Generally, the quantity of catalyst used will depend upon the nature of the catalyst, the reactor types and conditions and the properties desired for the polymer product. As is well known in the art hydrogen can be used for controlling the molecular weight of the polymer. The metallocene catalysts of the invention possess excellent catalyst activity and good comonomer response. The catalysts are also able to provide polymers of high weight average molecular weight Mw and narrow polydispersity Mw/Mn. Moreover, the random copolymerization behavior of metallocene catalysts of the invention shows a reduced tendency of chain transfer to ethylene. It is a feature of the invention that the claimed catalysts enable the formation of propylene polymers with high molecular weight. These features can be achieved at commercially interesting polymerization temperatures, e.g.60 °C or more, such as from 60 °C to 90 °C. The polydispersity index (Mw/Mn) of the polymers depend on the polymerization conditions
in each reactor, and can be between 2.0 and 7.0. In a particular embodiment, the propylene polymers obtained using the catalysts of the invention have a narrow polydispersity index (Mw/Mn), between 2.0 and 4.0. Propylene homopolymers Propylene homopolymers made by catalyst system comprising the metallocenes of the invention can be made with Mw (weight average molecular weight) values in the range of 40 to 2000 kg/mol, preferably in the range of 50 to 1500 kg/mol depending on the use and amount of hydrogen used as Mw regulating agent. The catalysts of the invention enable the formation of polypropylene homopolymers with high melting points. In a preferred embodiment the propylene homopolymer formed by the process of the invention has a melting point of more than 155 °C, preferably more than 157 °C. Propylene homopolymers having melting points up to 158 °C, or even up to 160 °C, are possible. Propylene copolymers Propylene copolymers with ethylene or with C4-C10 alpha olefin comonomers made by the metallocenes of the invention can be made with high productivity and low solubles. The polymers made by the catalysts of the description are useful in all kinds of end articles such as pipes, films (cast, blown or BOPP films, such as for example BOPP for capacitor film), fibers, moulded articles (e.g. injection moulded, blow moulded, rotomoulded articles), extrusion coatings and so on. The invention will now be illustrated by reference to the following non-limiting Examples. EXPERIMENTAL Measurement methods Al, B and Zr determination (ICP-method) In a glovebox, an aliquot of the catalyst (ca.40 mg) was weighed into glass weighting boat using analytical balance. The sample was then allowed to be exposed to air overnight while being placed in a steel secondary container equipped with an air intake. Then 5 mL of concentrated (65 %) nitric acid was used to rinse the content of the boat into the Xpress microwave oven vessel (20 mL). A sample was then subjected to a microwave-assisted digestion using MARS 6 laboratory microwave unit over 35 minutes at 150 °C. The digested sample was allowed to cool down for at least 4 h and then was transferred into a glass volumetric glass flask of 100 mL volume. Standard solutions containing 1000 mg/L Y and Rh (0.4 mL) were added. The flask was then filled up with distilled water and shaken
well. The solution was filtered through 0.45 µm Nylon syringe filters and then subjected to analysis using Thermo iCAP 6300 ICP-OES and iTEVA software. The instrument was calibrated for Al, B, Hf, Mg, Ti and Zr using a blank (a solution of 5 % HNO
3) and six standards of 0.005 mg/L, 0.01 mg/L, 0.1 mg/L, 1 mg/L, 10 mg/L and 100 mg/L of Al, B, Hf, Mg, Ti and Zr in solutions of 5 % HNO
3 distilled water. However, not every calibration point was used for each wavelength. Each calibration solution contained 4 mg/L of Y and Rh standards. Al 394.401 nm was calibrated using the following calibration points: blank, 0.1 mg/L, 1 mg/L, 10 mg/L and 100 mg/L. Al 167.079 nm was calibrated as Al 394.401 nm excluding 100 mg/L and Zr 339.198 nm using the standards of blank, 0.01 mg/L, 0.1 mg/L, 1 mg/L, 10 mg/L and 100 mg/L. Curvilinear fitting and 1/concentration weighting was used for the calibration curves. Immediately before analysis the calibration was verified and adjusted (instrument reslope function) using the blank and a 10 mg/L Al, B, Hf, Mg, Ti and Zr standard which had 4 mg/L Y and Rh. A quality control sample (QC: 1 mg/L Al, Au, Be, Hg & Se; 2 mg/L Hf & Zr, 2.5 mg/L As, B, Cd, Co, Cr, Mo, Ni, P, Sb, Sn & V; 4 mg/L Rh & Y; 5 mg/L Ca, K, Mg, Mn, Na & Ti; 10 mg/L Cu, Pb and Zn; 25 mg/L Fe and 37.5 mg/L Ca in a solution of 5 % HNO3 in distilled water) was run to confirm the reslope for Al, B, Hf, Mg, Ti and Zr. The QC sample was also run at the end of a scheduled analysis set. The content for Zr was monitored using Zr 339.198 nm {99} line. The content of aluminium was monitored via the 167.079 nm {502} line, when Al concentration in test portion was under 2 wt % and via the 394.401 nm {85} line for Al concentrations above 2 wt%. Y 371.030 nm {91} was used as internal standard for Zr 339.198 nm and Al 394.401 nm and Y 224.306 nm {450} for Al 167.079 nm. Catalyst Activity The catalyst activity was calculated on the basis of following formula: amount of polymer produced (kg) Catalyst Activity (kg-PP/g-Cat/h) = catalyst loading (g) × polymerisation time (h) The catalyst productivity was calculated on the basis of following formula: amount of polymer produced (kg) Catalyst productivity (kg-PP/g-Cat) = catalyst loading (g) Polymer powder bulk density Instruments: Electronic balance: Range from 0,1g-11000g
Graduated glass cylinder: Volume = max.250ml Plastic spoon: Volume=125ml Plastic funnel: D=105mm Execution: A glass cylinder is filled up to a volume of 250 ml by pouring in the unstabilised polymer powder, using a plastic spoon and a plastic funnel. Calculation: Mass of polymer (g)/measured volume (ml) XS The xylene soluble fraction (XS) was determined in line with ISO 16152 as follows: 2.5±0.1 g of the polymer were dissolved in 250 ml o-xylene under reflux conditions and continuous stirring, under nitrogen atmosphere. After 30 minutes, the solution was allowed to cool, first for 15 minutes at ambient temperature and then maintained for 30 minutes under controlled conditions at 25 ± 0.5 °C. The solution was filtered through filter paper. For determination of the xylene soluble content, an aliquot (100 ml) of the filtrate was taken. This aliquot was evaporated in nitrogen flow and the residue dried under vacuum at 100 °C until constant weight was reached. The xylene soluble fraction (weight percent) can then be determined as follows: XS% = (100 x m
1 x v
0)/(m
0 x v
1), wherein m
0 designates the initial polymer amount (grams), m
1 defines the weight of residue (grams), v
0 defines the initial volume (milliliter) and v
1 defines the volume of the analyzed sample (milliliter). To obtain the amorphous copolymer fraction for further characterization with GPC and NMR, the remaining xylene soluble filtrate was precipitated with acetone. The precipitated polymer was filtered and dried in the vacuum oven at 100 °C to constant weight. GPC: Molecular weight averages, molecular weight distribution, and polydispersity index (Mn, Mw, Mw/Mn) The MWD and the corresponded molecular weight averages Mn, Mw, Mv and Mz of the polymer sample were determined by using Gel Permeation Chromatography (GPC) at 160°C. All samples were integrated at the low Mw end up to the 3
rd last calibration point of the calibration curve (PS = 1820 g/mol ~ 1340 g/mol PP equivalent). A high temperature GPC equipped with a suitable concentration detector (like IR5 or IR4 from PolymerChar (Valencia, Spain), an online four capillary bridge viscometer (PL-BV 400-HT), and a dual light scattering detector (PL-LS 15/90 light scattering detector) with a 15° and 90° angle was used. 3x Olexis and 1x Olexis Guard columns from Agilent as
stationary phase and 1,2,4-trichlorobenzene (TCB, stabilized with 250 mg/L 2,6-Di tert butyl-4-methyl-phenol) as mobile phase at 160 °C and at a constant flow rate of 1 mL/min was applied. 200 μL of sample solution were injected per analysis. All samples were prepared by dissolving 8.0 – 10.0 mg of polymer in 10 mL (at 160 °C) of stabilized TCB (same as mobile phase) for 2,5 hours at 160°C under continuous gentle shaking. The injected concentration of the polymer solution at 160 °C (c
160°C) was determined in the following way. 0,8772
With: w
25 (polymer weight) and V
25 (Volume of TCB at 25°C). The column set was calibrated using universal calibration (according to ISO 16014-2:2019) with 19 narrow MWD polystyrene (PS) standards in the range of 0.5 kg/mol to 11500 kg/mol. The PS standards were dissolved at 160°C for 15 min or alternatively at room temperatures at a concentration of 0.2 mg/ml for molecular weight higher and equal 899 kg/mol and at a concentration of 1 mg/ml for molecular weight below 899 kg/mol. The conversion of the polystyrene peak molecular weight to polypropylene molecular weights was accomplished by using the Mark Houwink equation and the following Mark Houwink constants: K
PS = 19 x 10
-5 ml/g, α
PS = 0.655 K
PP = 39 x 10
-5 ml/g, α
PP = 0.725 A third order polynomial fit was used to fit the calibration data. All samples were prepared in the concentration range of 0.5 -1 mg/ml and dissolved at 160 °C for 3 hours under continuous gentle shaking Molecular weight averages (Mn, Mw, Mv and Mz), Molecular weight distribution (MWD) and its broadness, described by the polydispersity index PD= Mw/Mn (wherein Mn is the number average molecular weight and Mw is the weight average molecular weight) were determined using the following formulas:
The DSC curves and data have been produced on a DSC Q200 TA Instrument, by placing a 5-7 mg sample cut from the polymer MFR string, into a closed DSC aluminum pan, heating the sample from -10 °C to 225 °C at 10 °C/min, holding for 10 min at 225 °C, cooling from 225 °C to –30 °C, holding for 5 min at –30 °C, heating from –30 °C to 225 °C at 10 °C/min. The reported T
m values are those of the peak of the endothermic heat flow determined from the second heating scan. Melt Flow Rate The melt flow rate (MFR) was determined according to ISO 1133 and is indicated in g/10 min. The MFR is an indication of the flowability, and hence the processability, of the polymer. The higher the melt flow rate, the lower the molecular weight of the polymer. The MFR was determined at 230°C and may be determined at different loadings such as 2.16 kg (MFR2) or 21.6 kg (MFR21). Comonomer content by FTIR Quantitative infrared (IR) spectroscopy was used to estimate the C2 content of the copolymers through calibration to a primary method (NMR spectroscopy). Calibration was facilitated through the use of a set of in-house, non-commercial calibration standards of known C2 contents determined by quantitative
13C solution-state nuclear magnetic resonance (NMR) spectroscopy. The calibration procedure was undertaken in the conventional manner well documented in the literature [Spectroscopy of Polymers, 2nd Edition, J.L. Koenig, Elsevier Science, 1999]. The calibration set consisted of 8 calibration standards with C2 contents ranging between 0.0-3.5 wt%. Quantitative FTIR spectra were recorded in the solid-state using a Bruker Vertex 70 FTIR spectrometer. Spectra were recorded on 25x25 mm square films of 300 µm thickness prepared by compression moulding at 180 - 210°C and 70 bar of pressure. Standard transmission FTIR spectroscopy was employed using a spectral range of 5000-400 cm
-1, an aperture of 6 mm, a spectral resolution of 2 cm
-1, 16 background scans, 16 spectrum scans, an interferogram zero filling factor of 32 and Norton Beer strong apodisation.
Quantitative analysis was carried out by integration of (peak height) CH
2 rocking deformations at 732.5 cm
-1 (A
Q) corresponding to isolated ethylene incorporation in PEP comonomer sequences (integration method K-OPUS, limits 759 and 702 cm
-1). The quantitative band was normalized to the height of the CH combination band at 4323 cm
-1 (A
R) corresponding to CH structural units (integration method K, limits 4480, 3950 cm
-1). The C2 content in units of weight percent was then predicted from the normalized absorption (A
0 = A
Q / A
R) using linear calibration curve. The calibration curve having previously been constructed by least square regression of the normalized absorptions and comonomer contents measured by the primary technique (NMR Spectroscopy). The typical linear calibration curve has the form: ^ = ^
^ × ^
^ + ^
^ Equation 1 Here C
1 is the slope of the calibration curve with 0.96 > C
1 > 1, and the intercept of -0.02 > C
0 > 0.06. The quality of the calibration was assessed using the usual Confidence of Determination (COD) or R
2. The COD was around 0.998. NMR Quantitative nuclear-magnetic resonance (NMR) spectroscopy was used to quantify the isotacticity and content of regio-defects of the polypropylene homopolymers. Quantitative
13C{
1H} NMR spectra recorded in the solution-state using a Bruker Avance III 400 NMR spectrometer operating at 400.15 and 100.62 MHz for
1H and
13C respectively. All spectra were recorded using a
13C optimised 10 mm selective excitation probehead at 125°C using nitrogen gas for all pneumatics. Approximately 200 mg of material was dissolved in 1,2- tetrachloroethane-d
2 (TCE-d
2). This setup was chosen primarily for the high resolution needed for tacticity distribution quantification (Busico, V., Cipullo, R., Prog. Polym. Sci.26 (2001) 443; Busico, V.; Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). Standard single-pulse excitation was employed utilising the NOE and bi-level WALTZ16 decoupling scheme (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, B., J. Mag. Reson.187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28, 11289). A total of 6144 (6k) transients were acquired per spectra using a 3 s recycle delay. Quantitative
13C{
1H} NMR spectra were processed, integrated and relevant quantitative properties determined from the integrals using proprietary computer programs. All chemical shifts are internally referenced to the methyl signal of the isotactic pentad mmmm at 21.85 ppm.
The tacticity distribution was quantified through integration of the methyl region between 23.6 and 19.7 ppm correcting for any sites not related to the stereo sequences of interest (Busico, V., Cipullo, R., Prog. Polym. Sci.26 (2001) 443; Busico, V., Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). The pentad isotacticity was determined through direct integration of the methyl region and reported as either the mole fraction or percentage of isotactic pentad mmmm with respect to all steric pentads i.e. [mmmm] = mmmm / sum of all steric pentads. When appropriate integrals were corrected for the presence of sites not directly associated with steric pentads. Characteristic signals corresponding to regio irregular propene insertion were observed (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253). The presence of secondary inserted propene in the form of 2,1 erythro regio defects was indicated by the presence of the two methyl signals at 17.7 and 17.2 ppm and confirmed by the presence of other characteristic signals. The amount of 2,1 erythro regio defects was quantified using the average integral (e) of the e6 and e8 sites observed at 17.7 and 17.2 ppm respectively, i.e. e = 0.5 * (e6 + e8). Characteristic signals corresponding to other types of regio irregularity were not observed (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253). The amount of primary inserted propene (p) was quantified based on the integral of all signals in the methyl region (CH3) from 23.6 to 19.7 ppm paying attention to correct for other species included in the integral not related to primary insertion and for primary insertion signals excluded from this region such that p = CH3 + 2*e. The relative content of a specific type of regio defect was reported as the mole fraction or percentage of said regio defect with respect all observed forms of propene insertion i.e. sum of all primary (1,2), secondary (2,1) and tertiary (3,1) inserted propene units, e.g. [21e] = e / ( p + e + t + i ). The total amount of secondary inserted propene in the form of 2,1-erythro or 2,1-threo regio defects was quantified as sum of all said regio irregular units, i.e. [21] = [21e] + [21t]. Quantitative nuclear-magnetic resonance (NMR) spectroscopy was used to quantify the ethylene content and the isotacticity of the copolymers. Quantitative
13C{
1H} NMR spectra were recorded in the solution-state using a Bruker Avance III 400 NMR spectrometer operating at 400.15 and 100.62 MHz for
1H and
13C respectively. All spectra were recorded using a
13C optimised 10 mm extended temperature probehead at 125°C using nitrogen gas for all pneumatics. Approximately 200 mg of material was dissolved in 3 ml of 1,2-tetrachloroethane-d2 (TCE-d2) along with chromium-(III)-acetylacetonate (Cr(acac)
3) resulting in a 65 mM solution of relaxation
agent in solvent as described in G. Singh, A. Kothari, V. Gupta, Polymer Testing 2009, 28(5), 475. To ensure a homogenous solution, after initial sample preparation in a heat block, the NMR tube was further heated in a rotatory oven for at least 1 hour. Upon insertion into the magnet the tube was spun at 10 Hz. This setup was chosen primarily for the high resolution and quantitatively needed for accurate ethylene content quantification. Standard single- pulse excitation was employed without NOE, using an optimised tip angle, 1 s recycle delay and a bi-level WALTZ16 decoupling scheme as described in Z. Zhou, R. Kuemmerle, X. Qiu, D. Redwine, R. Cong, A. Taha, D. Baugh, B. Winniford, J. Mag. Reson.187 (2007) 225 and V. Busico, P. Carbonniere, R. Cipullo, C. Pellecchia, J. Severn, G. Talarico, Macromol. Rapid Commun.2007, 28, 1128. A total of 6144 (6k) transients were acquired per spectra. Quantitative
13C{
1H} NMR spectra were processed, integrated and relevant quantitative properties determined from the integrals. All chemical shifts were indirectly referenced to the central methylene group of the ethylene block (EEE) at 30.00 ppm using the chemical shift of the solvent. This approach allowed comparable referencing even when this structural unit was not present. With characteristic signals corresponding to 2,1 erythro regiodefects observed (as described in L. Resconi, L. Cavallo, A. Fait, F. Piemontesi, Chem. Rev. 2000, 100 (4), 1253, in Cheng, H. N., Macromolecules 1984, 17, 1950, and in W-J. Wang and S. Zhu, Macromolecules 2000, 33, 1157) the correction for the influence of the regiodefects on determined properties was required. Characteristic signals corresponding to other types of regiodefects were not observed. Characteristic signals corresponding to the incorporation of ethylene were observed (as described in Cheng, H. N., Macromolecules 1984, 17, 1950) and the comonomer fraction calculated as the fraction of ethylene in the polymer with respect to all monomer in the polymer: fE = ( E / ( P + E ) The comonomer fraction was quantified using the method of W-J. Wang and S. Zhu, Macromolecules 2000, 33, 1157, through integration of multiple signals across the whole spectral region in the
13C{
1H} spectra. This method was chosen for its robust nature and ability to account for the presence of regio-defects when needed. Integral regions were slightly adjusted to increase applicability across the whole range of encountered comonomer contents. The mole percent comonomer incorporation was calculated from the mole fraction:
E [mol%] = 100 * fE The weight percent comonomer incorporation was calculated from the mole fraction: E [wt%] = 100 * ( fE * 28.06 ) / ( (fE * 28.06) + ((1-fE) * 42.08) ) The isotacticity of the copolymer was determined according to known methods, for example as described in Macromolecules 2005, vol.38, pp.3054-3059. Crystex The crystalline (CF) and soluble fractions (SF) of the heterophasic propylene resins as well as the comonomer content and intrinsic viscosities of the respective fractions were analysed by the Crystex method. The crystalline and amorphous fractions are separated through temperature cycles of dissolution at 160°C, crystallization at 40°C and re- dissolution in 1,2,4-trichlorobenzene (1,2,4-TCB) at 160°C. Quantification of SF and CF and determination of ethylene content (C2) were achieved by means of an infrared detector (IR4) and an online 2-capillary viscometer was used for determination of the intrinsic viscosity (iV). IR4 detector is multiple wavelength detector detecting IR absorbance at two different bands (CH3 and CH2) for the determination of the concentration and the ethylene content in ethylene-propylene copolymers. IR4 detector is calibrated with series of EP copolymers with known ethylene content in the range of 2 wt.-% to 69 wt.-% (determined by 13C- NMR). Amount of Soluble fraction (SF) and Crystalline Fraction (CF) are correlated through the XS calibration to the “Xylene Soluble” (XS) quantity and respectively Xylene Insoluble (XI) fractions, determined according to standard gravimetric method as per ISO16152 (2005). XS calibration is achieved by testing various EP copolymers with XS content in the range 2-31 wt%. Intrinsic viscosity (iV) of the parent EP copolymer and its soluble and crystalline fractions are determined with a use of an online 2-capillary viscometer and are correlated to corresponding iV determined in decalin according to ISO 1628-3 (2010). Calibration is achieved with several commercial EP PP copolymers with iV = 2-4 dL/g. A sample of the PP composition to be analysed is weighed out in concentrations of 10mg/ml to 20mg/ml. After automated filling of the vial with 1,2,4-TCB containing 250 mg/l 2,6-tert-butyl-4-methylphenol (BHT) as antioxidant, the sample is dissolved at 160°C until complete dissolution is achieved, usually for 60 min, with constant stirring of 800rpm. A defined volume of the sample solution is injected into the column filled with inert support where the crystallization of the sample and separation of the soluble fraction from the
crystalline part is taking place. This process is repeated two times. During the first injection the whole sample is measured at elevated temperature, determining the iV[dl/g] and the C2[wt%] of the PP composition. During the second injection the soluble fraction (at low temperature) and the crystalline fraction (at elevated temperature) with the crystallization cycle are determined (wt% SF, wt% C2, iV). Metallocene synthesis Synthesis of MC-CE1 MC-CE1 was prepared as described in WO2001048034, metallocene example 18. Synthesis of MC-CE2 MC-CE2, was prepared as described in WO2005058916, metallocene example 1. Synthesis of MC-CE3 Rac-dimethylsilanediyl-bis[2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert- butyl-inden- l-yl]zirconium dichloride, MC-CE3, was prepared as described in WO2018091684 for MC- IE1. Synthesis of MC-CE4 Rac-dimethylsilanediyl-bis[2-neopentyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert- butylinden-1–yl]zirconium dichloride, was prepared as described in WO2018091684 for MC-IE4. Synthesis of MC-IE1 and MC-IE2 Isopropylmalonic acid
A solution of 125 g of potassium hydroxide in 1000 cm
3 of water was added to a solution of 110.0 g (544 mmol) of diethyl isopropylmalonate in 500 ml of methanol. The resulting mixture was refluxed for 5 h, then ethanol and methanol were distilled off. Then 1000 cm
3 of water was added, and the obtained mixture was acidified by 12 M HCl to pH 1.0. Isopropylmalonic acid was extracted with 4 ^500 ml of ether. The combined extract was evaporated to dryness, and the residue was dried in vacuum. This procedure gave 76.4 g (96.1%) of isopropylmalonic acid as white solid.
1H NMR (CDCl
3): δ 9.72 (br.s, 2H), 3.24 (d, J = 8.3 Hz, 1H), 2.48-2.34 (m, 1H), 1.07 (d, J = 6.8 Hz, 6H). 2-Isopropylacrylic acid
Diethylamine (62.4 ml, 4.3 g, 0.606 mol) was added dropwise at 5 °C to a solution of isopropylmalonic acid (76.4 g, 523 mmol) in 750 ml of ethyl acetate. Paraform (22.1 g, 0.736 mol) was added to the obtained suspension. The resulting mixture was refluxed for 5 h, then cooled to 5 °C, and 350 ml of ether and 1000 cm
3 of 2 M HCl were added. After mixing, the organic layer was separated, the aqueous layer was additionally extracted with 2 ^500 ml of ether. The combined organic phase was dried over Na2SO4 and then evaporated to dryness. The residue was purified by vacuum distillation to give 2- isopropylacrylic acid, bp 65 °C/4 mm Hg. Yield 57.0 g (95.5%) of a colorless liquid.
1H NMR (CDCl
3): δ 12.45 (br.s, 1H), 6.30 (s, 1H), 5.65 (t, 1H), 2.81 (septd, J = 6.9 Hz, J = 0.9 Hz, 1H), 1.11 (d, J = 6.9 Hz, 6H).
13C NMR (CDCl
3): δ 173.30, 146.47, 124.31, 28.94, 21.76. 6-tert-Butyl-5-methoxy-2-isopropylindan-1-one
114.1 g (1.0 mol) of 2-isopropylacrylic acid was added to Eaton's reagent obtained from 220 g of P4O10 and 1120 ml of MeSO3H at 50 °C. 131.2 g (0.8 mol) of 1-tert-butyl-2- methoxybenzene was added dropwise to this mixture with vigorous stirring for ca.1 h at 50-53 °C (water bath temperature). The resulting mixture was stirred for 1 h at this temperature, then cooled to room temperature, and poured on a mixture of 1.5 liter of cold water and 3 kg of ice. The crude product was extracted with 3 ^600 ml of dichloromethane. The combined organic phase was washed by aqueous K2CO3, dried over K2CO3, filtered through a short pad of silica gel 60 (40-63 µm) and then evaporated to dryness. The residue was purified by vacuum distillation to give 193.8 g (93.0 %, ca.95% purity) of 6- tert-butyl-5-methoxy-2-isopropylindan-1-one as a yellowish oil (bp 150-190
oC /4 mm Hg).
1H NMR (CDCl
3): δ 7.66 (s, 1H), 6.89 (s, 1H), 3.93 (s, 3H), 3.04 (dd, J = 17.4 Hz, J = 8.0 Hz, 1H), 2.84 (dd, J = 17.4 Hz, J = 3.8 Hz, 1H), 2.67-2.60 (m, 1H), 2.48-2.34 (m, 1H), 1.37 (s, 9H), 1.05 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.9 Hz, 3H).
13C NMR (CDCl
3): δ 207.45, 164.44, 155.10, 138.54, 130.07, 121.69, 107.64, 55.14, 53.20, 35.00, 29.54, 28.87, 27.65, 20.96, 17.00. 4-Bromo-6-tert-butyl-2-isopropyl-5-methoxyindan-1-one
Bromine (20.8 ml, 64.9 g, 405.9 mmol) was added dropwise over 5 min to a mixture of 97.0 g (0.372 mol) of 6-tert-butyl-2-isopropyl-5-methoxyindan-1-one, 113.2 g of sodium acetate, 3.0 g of
nBu4NI, 310 ml of dichloromethane, and 645 ml of water with vigorous stirring at 5 °C. This mixture was stirred at 5 °C for 2 h, then a solution of 52.2 g of sodium acetate in 290 ml of water was added followed by addition of 10.8 ml (33.7 g, 210.8 mmol) of bromine. The resulting mixture was additionally stirred for 1 h at this temperature and then washed by aqueous Na2SO3 to remove excess bromine. The crude product was extracted by 3 ^250 ml of dichloromethane. The combined organic extract was dried over K2CO3, evaporated to dryness, and the residue was dried in vacuum. This procedure gave 124.84 g (98.9%, ca. 95% purity) of a yellowish oil which was used without further purification.
1H NMR (CDCl3): δ 7.68 (s, 1H), 4.04 (s, 3H), 3.04 (dd, J = 17.9 Hz, J = 8.1 Hz, 1H), 2.80 (dd, J = 17.9 Hz, J = 3.9 Hz, 1H), 2.71-2.64 (m, 1H), 2.49-2.35 (m, 1H), 1.40 (s, 9H), 1.08 (d, J = 6.9 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H).
13C NMR (CDCl3): δ 207.21, 162.59, 154.47, 145.24, 133.82, 121.03, 116.56, 61.54, 53.32, 35.56, 30.54, 29.41, 28.94, 20.78, 17.17. 6-tert-Butyl-2-isopropyl-5-methoxy-4-(3,5-dimethylphenyl)-indan-1-one
A mixture of 124.84 g (368.0 mmol) of 4-bromo-6-tert-butyl-2-isopropyl-5-methoxyindan- 1-one, 69.7 g (464.7 mmol, 1.26 equiv.) of 3,5-Me
2C
6H
3B(OH)
2, 1.9 g (3.72 mmol, 1 mol. %) of Pd(P
tBu
3)
2, 118.3 g of Na
2CO
3, 600 ml of 2-methyltetrahydrofurane and 540 ml of water was refluxed for 6 h. Then, 500 ml of water was added, the organic layer was separated, and the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic extract was dried over K
2CO
3 and then evaporated to dryness to give slightly yellowish solid mass. The product was isolated by flash-chromatography on silica gel 60 (40-63 µm, eluent: hexanes-dichloromethane = 1:1 and then 1:5, vol.). Yield 121.55 g (90.6%, purity ca.95%) of a yellowish crystalline material.
1H NMR (CDCl
3): δ 7.71 (s, 1H), 7.04 (s, 1H), 7.03 (s, 2H), 3.31 (s, 3H), 2.87 (dd, J = 18.5 Hz, J = 8.8 Hz, 1H), 2.65-2.54 (dd and m, 2H), 2.43-2.34 (s and m, 7H), 1.42 (s, 9H), 0.99 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.8 Hz, 3H).
13C NMR (CDCl
3): δ 208.19, 163.34, 153.44, 143.11, 138.08, 136.33, 132.64, 132.12, 129.08, 127.20, 120.93, 77.00, 60.46, 53.37, 35.33, 30.50, 28.88, 27.38, 21.39, 20.90, 17.25. 5-tert-Butyl-2-isopropyl-6-methoxy-7-(3,5-dimethylphenyl)-1H-indene
NaBH
4 (18.9 g, 0.5 mol, 1.5 equiv.) was added to a solution of 121.55 g (333.45 mmol) of 6-tert-butyl-2-isopropyl-5-methoxy-4-(3,5-dimethylphenyl)-indan-1-one in 600 ml of THF cooled to 5 °C. MeOH (300 ml) was added dropwise to this mixture over ca.5 h at 5 °C, and the resulting mixture was stirred overnight at room temperature. Then, this mixture was evaporated to dryness, 1000 ml of dichloromethane and 1000 ml water were added to the residue, and the so obtained mixture was acidified with 2 M HCl to pH~6.5. The organic layer was separated, the aqueous layer was additionally extracted with 100 ml of dichloromethane. The combined organic phase was passed through a pad (~30 ml) of silica gel 60 (40-63 µm; eluent: dichloromethane) to get rid of most of the palladium black. The obtained elute was evaporated to dryness to give a grey solid mass. To a solution of this mass in 1000 ml of toluene 1.0 g of TsOH was added. This mixture was refluxed with Dean-Stark head for 10 min and then cooled to room temperature using water bath. The formed solution was washed with 10% Na2CO3, the organic layer was separated, the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic
phase was dried over K
2CO
3 and then evaporated to dryness. The crude product was purified by flash chromatography on silica gel 60 (40-63 µm, hexanes-dichloromethane = 5:1) followed by recrystallization from n-hexane (hot→ –30 °C). This procedure gave 96.93 g (83.4%) of pure 5-tert-butyl-7-(3,5-dimethylphenyl)-2-isopropyl-6-methoxy-1H-indene.
1H NMR (CDCl
3): δ 7.24 (s, 1H), 7.09 (s, 2H), 6.99 (s, 1H), 6.46 (m, 1H), 3.24 (s, 3H), 3.15 (m, 2H), 2.68 (sept, J = 6.8 Hz, 1H), 2.37 (s, 6H), 1.43 (s, 9H), 1.14 (d, J = 6.8 Hz, 6H).
13C NMR (CDCl
3): δ 156.43, 154.33, 141.34, 140.95, 140.23, 138.29, 137.69, 131.99, 128.49, 127.22, 123.86, 117.32, 60.67, 39.15, 35.13, 31.00, 30.04, 22.64, 21.44. [6-tert-Butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-methyl-1H-inden-1-yl] chlorodimethylsilane
nBuLi in hexanes (2.43 M, 14.6 ml, 35.5 mmol) was added in one portion to a solution of 5-tert-butyl-7-(3,5-dimethylphenyl)-6-methoxy-2-methyl-1H-indene (11.3 g, 35.3 mmol) in 200 ml of ether cooled to –50 °C. The resulting orange solution was stirred overnight at room temperature, then the obtained orange solution containing a yellowish precipitate was cooled to –78 °C (the precipitate almost completely disappeared), and dichlorodimethylsilane (22.8 g, 177 mmol, 5 equiv) was added in one portion. The formed solution was warmed to room temperature and stirred overnight at room temperature. The resulting mixture was filtered through glass frit (G4). The precipitate was additionally washed by 2×10 ml of ether. The combined filtrate was evaporated to dryness to give the title material as slightly orange oil which was used without further purification.
anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1- yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]hafnium dichloride
nBuLi in hexanes (2.5 M, 8.0 ml, 20.0 mmol) was added in one portion at room temperature to a yellowish solution of [6-tert-butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-isopropyl-1H- inden-1-yl][6-tert-butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-methyl-1H-inden-1-yl] dimethylsilane (7.25 g, ca.10.0 mmol) in 50 ml of
nBu2O. This mixture was stirred overnight at room temperature, then the resulting red solution was cooled to 0 °C in an ice-bath, and HfCl4 (3.2 g, 9.99 mmol) was added. The reaction mixture was stirred for 24 h at room temperature to give a red solution with precipitate of LiCl. This mixture was evaporated to dryness, and the residue was extracted with 4 ^30 ml of hot n-hexane. The combined extract was evaporated to ca.10 ml, then 20 ml of n-pentane was added to the resulting solution. The yellow precipitate fallen from this solution overnight at room temperature was filtered off and dried in vacuum. This procedure gave 1.45 g of a ca.1:1 mixture of syn- and anti-complexes containing 1 equiv. of n-pentane (or 0.1 g of n-pentane in 1.45 g of the product), so the adjusted net weight of the isolated anti/syn mixture was 1.35 g. The mother liquor was evaporated to dryness, and the residue was dissolved in 30 ml of n-pentane. The yellow precipitate fallen from this solution for 2 days at room temperature was filtered off and dried in vacuum. This procedure gave 3.7 g of pure anti-complex. Thus, the total yield of syn- and anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy--6- tert-butylinden-1-yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl] hafnium dichlorides isolated in this synthesis was found to be 5.05 g (51.9%).
Anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy--6-tert-butylinden- 1-yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl] hafnium dichloride. Anal. calc. for C
50H
62Cl
2HfO
2Si: C, 61.75; H, 6.43. Found: C, 62.01; H, 6.64.
1H NMR (CDCl
3): δ 7.64-6.87 (very br.s, 4H), 7.55 (s, 1H), 7.49 (s, 1H), 6.98 (s, 1H), 6.96 (s, 1H), 6.53 (s, 1H), 6.48 (s, 1H), 3.43 (s, 3H), 3.41 (s, 3H), 3.30 (sept, J = 6.7 Hz, 1H), 2.36 (s, 6H), 2.35 (s, 6H), 2.24 (s, 3H), 1.39 (s, 18H), 1.28 (s, 6H), 1.10 (d, J = 6.8 Hz, 3H), 1.03 (d, J = 6.6 Hz, 3H). Synthesis of MC-IE3 7-Methyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one Methacryloyl chloride (94.39 g, 903.0 mmol) was added dropwise over 15 min to a suspension of AlCl
3 (126.4 g, 947.7 mmol) in 750 ml of dichloromethane cooled to –78 °C a, followed by dropwise addition of benzo-1,4-dioxane (123.0 g, 903.4 mmol). The reaction mixture was heated to room temperature in 1 hour, then the reaction mixture was stirred for 19 h at room temperature. The resulting mixture was poured onto 2000 cm
3 of crushed ice. The organic layer was separated, the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic extract was washed with aqueous K
2CO
3, dried over K
2CO
3, and passed through a short pad of silica gel 60 (40-63 µm) which was additionally washed with 200 ml of dichloromethane. The combined organic elute was evaporated to dryness to give 167.5 g (90.8%, purity ca. 90%) of 7-methyl-2,3,7,8- tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one as a white solid mass which was used without further purification. 9-Bromo-7-methyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one
A mixture of 93.3 g (456.9 mmol) of 7-methyl-2,3,7,8-tetrahydro-6H-indeno[5,6- b][1,4]dioxin-6-one, 139.0 g of NaOAc, 3.5 g of
nBu
4NBr, 400 ml of dichloromethane, and 800 ml of water was cooled to +5 °C, then bromine (23.5 ml, 73.3 g, 458.7 mmol) was added dropwise over 20 min at this temperature. The resulting mixture was stirred for 1 h
at this temperature, then a solution of 63.6 g of NaOAc in 400 ml of water was added followed by 11.0 ml (34.3 g, 214.7 mmol) of bromine for 10 min. This mixture was stirred additionally for 1 h at 5 °C, then washed by aqueous Na
2SO
3 to neutralize any excess bromine. The organic layer was separated, and the aqueous phase was additionally extracted with 2 ^200 ml of dichloromethane. The combined organic extract was filtered through a pad of silica gel 60 (40-63 µm) which was additionally washed with 200 ml of dichloromethane. The combined organic elute was evaporated to ca.200 ml, and 200 ml of n-hexane was added. The precipitated white solid was filtered off (G3) and dried under vacuum to give 56.56 g (43.7%) of pure 9-bromo-7-methyl-2,3,7,8-tetrahydro-6H- indeno[5,6-b][1,4]dioxin-6-one. The mother liquor was evaporated to give a semi-solid residue. This residue was triturated with 65 ml of dichloromethane, then 65 ml of n-hexane was added. The precipitated white solid was filtered off (G3) and dried under vacuum to give 34.54 g of a 2:1 mixture of 9-bromo-7-methyl-2,3,7,8-tetrahydro-6H-indeno[5,6- b][1,4]dioxin-6-one and 5,9-dibromo-7-methyl-2,3,7,8-tetrahydro-6H-indeno[5,6- b][1,4]dioxin-6-one, respectively.
1H NMR (CDCl
3): δ 7.22 (s, 1H), 4.47-4.42 (m, 2H), 4.32- 4.27 (m, 2H), 3.26 (dd, J = 17.3 Hz, J = 7.8 Hz, 1H), 2.70 (dqd, J = 7.8 Hz, J = 7.5 Hz, J = 3.8 Hz, 1H), 2.57 (dd, J = 17.3 Hz, J = 3.8 Hz, 1H), 1.30 (d, J = 7.5 Hz, 3H).
13C NMR (CDCl
3): δ 207.52, 147.15, 146.65, 144.14, 130.05, 110.79, 108.95, 65.28, 63.60, 41.92, 35.34, 16.36. 9-(3,5-Dimethylphenyl)-7-methyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6- one
A mixture of 55.48 g (199.49 mmol) of 9-bromo-7-methyl-2,3,7,8-tetrahydro-6H- indeno[5,6-b][1,4]dioxin-6-one, 37.84 g (252.3 mmol, 1.27 equiv.) of 3,5-dimethylphenyl- boronic acid, 1.04 g (2.04 mmol, 1 mol.%) of Pd(P
tBu3)2, 64.3 g of Na2CO3, 330 ml of 2- methyltetrahydrofurane, and 295 ml of water was refluxed for 2 h. Further on, 400 ml of water was added, the organic layer was separated, and the aqueous layer was extracted with 400 ml of dichloromethane. The combined organic extract was dried over K2CO3 and then evaporated to dryness to give a light brown solid mass. The product was isolated by flash-chromatography on silica gel 60 (40-63 µm, eluent: dichloromethane, then dichloromethane:ether = 1:10, vol.). The product containing fractions were combined and evaporated to dryness. The resulting solid mass was triturated with 200 ml of n-hexane.
The formed precipitate was filtered off (G3), washed with 2 ^40 ml of n-hexane, and dried under vacuum. This procedure gave 59.5 g (96.7%) of 9-(3,5-dimethylphenyl)-7-methyl- 2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one as a white powder.
1H NMR (CDCl
3): δ 7.27 (s, 1H), 7.03 (br.s, 1H), 6.95 (br.s, 2H), 4.30-4.23 (m, 4H), 3.10 (dd, J = 17.1 Hz, J = 7.8 Hz, 1H), 2.61 (dqd, J = 7.8 Hz, J = 7.6 Hz, J = 3.9 Hz, 1H), 2.42 (dd, J = 17.1 Hz, J = 3.9 Hz, 1H), 2.38 (s, 6H), 1.24 (d, J = 7.6 Hz, 3H).
13C NMR (CDCl3): δ 208.42, 146.77, 146.02, 143.90, 137.76, 134.19, 129.42, 129.21, 128.28, 127.15, 110.67, 64.72, 63.59, 41.97, 33.93, 21.35, 16.37. 5-(3,5-Dimethylphenyl)-7-methyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxine
NaBH4 (11.0 g, 290.8 mmol, 1.5 equiv.) was added to a solution of 9-(3,5-dimethylphenyl)- 7-methyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one (59.5 g, 193.0 mmol) in 550 ml of THF cooled to 5 °C. To this mixture, 200 ml of MeOH was added dropwise over ca. 5 h at 5 °C, and the resulting mixture was stirred overnight at room temperature. Then, this mixture was evaporated to dryness, 1000 ml of dichloromethane and 1000 ml water were added to the residue, and the so obtained mixture was acidified with 2 M HCl to pH~6.5. The organic layer was separated, the aqueous layer was additionally extracted with 50 ml of dichloromethane. The combined organic extract was passed through a pad (~30 ml) of silica gel 60 (40-63 µm; eluent: dichloromethane) to get rid of most of the palladium black. The obtained elute was evaporated to dryness to give a white solid mass which was dissolved in 800 ml of toluene, preheated to ca.60 °C, then TsOH (1.0 g) was added. This mixture was refluxed with Dean-Stark head for 10 min. Then, the reaction mixture was quickly cooled to room temperature using an ice-water bath. The formed solution was washed with 10% aqueous K
2CO
3, the organic layer was separated, the aqueous layer was extracted with 100 ml of dichloromethane. The combined organic extract was dried over K
2CO
3, passed through a pad of silica gel 6040-63 µm), and the so obtained elute was evaporated to dryness. The crude product was triturated with 200 ml of n-hexane. The formed precipitate was filtered off (G3), then washed with 2x20 ml of n-hexane, and dried under vacuum. This procedure gave 53.08 g (96.7%) of 5-(3,5-dimethylphenyl)-7-methyl- 2,3-dihydro-6H-indeno[5,6-b][1,4]dioxine as a white powder.
1H NMR (CDCl
3): δ 7.01 (br.s, 2H), 6.98 (br.s, 1H), 6.76 (s, 1H), 6.37 (m, 1H), 4.24-4.21 (m, 2H), 4.21-4.17 (m, 2H), 3.08 (s, 2H), 2.36 (s, 6H), 2.05 (m, 3H).
13C NMR (CDCl
3): δ 145.32, 142.45, 138.59, 137.46,
136.35, 135.27, 128.94, 127.26, 127.18, 126.28, 107.50, 64.40, 64.15, 42.18, 21.41, 16.66. 7-Isopropyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one
2-isopropylacryloyl chloride (78.0 g, 588 mmol) was added dropwise over 15 min to a suspension of 82.3 g (617 mmol) of AlCl
3 in 500 ml of dichloromethane cooled to –78 °C, followed by dropwise addition of benzo-1,4-dioxane (80.05 g, 588 mmol). The temperature of the reaction mixture was raised to room temperature in 1 h, then the reaction mixture was stirred for 19 h at room temperature. The resulting mixture was poured onto 1500 cm
3 of crushed ice. The organic layer was separated, the aqueous layer was extracted with 2x200 ml of dichloromethane. The combined organic extract was washed with aqueous K2CO3, dried over K2CO3 and passed through a short pad of silica gel 60 (40-63 µm), which was additionally washed with 200 ml of dichloromethane. The combined organic elute was evaporated to dryness. The residue was washed with 300 ml of n-hexane and dried under vacuum to give 105.4 g (77.1%) of the title material as a white solid mass.
1H NMR (CDCl3): δ 7.22 (s, 1H), 6.90 (s, 1H), 4.33-4.31 (m, 2H), 4.27-4.25 (m, 2H), 3.00 (dd, J = 17.1 Hz, J = 8.0 Hz, 1H), 2.79 (dd, J = 17.1 Hz, J = 3.8 Hz, 1H), 2.64-2.60 (m, 1H), 2.42-2.32 (m, 1H), 1.03 (d, J = 6.9 Hz, 3H), 0.76 (d, J = 6.8 Hz, 3H).
13C NMR (CDCl3): δ 207.40, 149.83, 148.14, 143.50, 131.19, 113.89, 111.37, 64.60, 63.82, 53.31, 29.04, 27.39, 20.86, 17.02. 9-Bromo-7-isopropyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one
A mixture of 51.1 g (220 mmol) of 7-isopropyl-2,3,7,8-tetrahydro-6H-indeno[5,6- b][1,4]dioxin-6-one, 67.0 g of NaOAc, 1.8 g of
nBu4NBr, 200 ml of dichloromethane, and 400 ml of water was cooled to +5 °C, and then bromine (11.3 ml, 35.15 g, 220 mmol) was added dropwise over 20 min at this temperature. The resulting mixture was stirred for 1 h at this temperature, then a solution of 16.4 g of NaOAc in 100 ml of water was added, and finally bromine (2.6 ml, 8.0 g, 50 mmol) was added over 10 min. This mixture was stirred for 1 h at 5 °C, then the resulting mixture was washed with aqueous Na
2SO
3 to remove excess bromine. The organic layer was separated, and the aqueous layer was additionally
extracted with 2x200 ml of dichloromethane. The combined organic extract was filtered through a pad of silica gel 60 (40-63 µm), which was additionally washed with 200 ml of dichloromethane. The combined organic elute was evaporated to dryness. The crude product was purified by flash chromatography on silica gel 60 (40-63 µm, d 50 mm, l 1000 mm, eluent: dichloromethane). Yield 37.2 g (54.3%).
1H NMR (CDCl
3): δ 7.20 (s, 1H), 4.46- 4.44 (m, 2H), 4.30-4.28 (m, 2H), 3.00 (dd, J = 17.5 Hz, J = 8.0 Hz, 1H), 2.75 (dd, J = 17.5 Hz, J = 3.8 Hz, 1H), 2.66 (m, 1H), 2.39 (m, 1H), 1.05 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.8 Hz, 3H).
13C NMR (CDCl
3): δ 206.84, 147.82, 146.57, 144.09, 131.34, 110.44, 108.98, 65.29, 63.61, 53.07, 29.05, 28.79, 20.73, 17.06. 9-(3,5-Dimethylphenyl)-7-isopropyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6- one
A mixture of 37.2 g (119.6 mmol) of 9-bromo-7-isopropyl-2,3,7,8-tetrahydro-6H- indeno[5,6-b][1,4]dioxin-6-one, 22.78 g (151.9 mmol, 1.27 equiv.) of 3,5- dimethylphenylboronic acid, 0.61 g (1.2 mmol, 1 mol.%) of Pd(P
tBu
3)
2, 38.5 g of Na
2CO
3, 200 ml of 2-methyltetrahydrofurane, and 180 ml of water was refluxed for 2 h. Then, 400 ml of water was added, the organic layer was separated, and the aqueous layer was extracted with 2x200 ml of dichloromethane. The combined organic extract was dried over K2CO3 and then evaporated to dryness. The product was isolated by flash chromatography on silica gel 60 (40-63 µm, d 50 mm, l 500 mm, eluent: dichloromethane, then, dichloromethane:ether = 1:10, vol.). This procedure gave 37.4 g (93%) of the title product as a white powder.
1H NMR (CDCl3): δ 7.26 (s, 1H), 7.05 (br.s, 1H), 6.96 (br.s, 2H), 4.30- 4.23 (m, 4H), 2.85 (dd, J = 18.1 Hz, J = 8.6 Hz, 1H), 2.60-2.54 (m, 2H), 2.39 (s, 6H), 2.39- 2.32 (m, 1H), 0.97 (d, J = 6.9 Hz, 3H), 0.75 (d, J = 6.8 Hz, 3H).
13C NMR (CDCl3): δ 207.75, 146.72, 143.83, 137.83, 134.36, 130.59, 129.46, 128.30, 127.19, 110.35, 64.74, 63.61, 53.12, 29.02, 27.09, 21.39, 20.83, 17.15. 5-(3,5-Dimethylphenyl)-7-isopropyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxine
NaBH
4 (6.35 g, 168.0 mmol, 1.5 equiv.) was added to a solution of 9-(3,5-dimethylphenyl)- 7-isopropyl-2,3,7,8-tetrahydro-6H-indeno[5,6-b][1,4]dioxin-6-one (37.4 g, 111.2 mmol) in 380 ml of THF cooled to 5 °C. To this mixture, 140 ml of MeOH was added dropwise over ca.5 h at 5 °C, and the resulting mixture was stirred overnight at room temperature. Then, this mixture was evaporated to dryness, 500 ml of dichloromethane and 1000 ml water were added to the residue, and the so obtained mixture was acidified with 2 M HCl to pH~6.5. The organic layer was separated, the aqueous layer was additionally extracted with 2x50 ml of dichloromethane. The combined organic extract was evaporated to dryness to give a white solid mass, which was dissolved in 500 ml of toluene, preheated to ca.60 °C, then 1.0 g of TsOH was added. This mixture was refluxed with Dean-Stark head for 10 min. Then, the reaction mixture was quickly cooled to room temperature using an ice-water bath. The formed solution was washed with 10% K
2CO
3, the organic layer was separated, and the aqueous layer was extracted with 100 ml of dichloromethane. The combined organic extract was dried over K2CO3, passed through a pad of silica gel 60 (40- 63 µm), and then evaporated to dryness. The crude product was triturated with 50 ml of n- hexane, the precipitate was filtered off (G3), the filter cake was washed with 20 ml of n- hexane and dried under vacuum. This procedure gave 31.8 g (89%) of the title product as a white powder.
1H NMR (CDCl3): δ 7.02 (br.s, 2H), 6.99 (br.s, 1H), 6.79 (s, 1H), 6.38 (m, 1H), 4.22-4.20 (m, 2H), 4.18-4.16 (m, 2H), 3.11 (s, 2H), 2.66 (sep, J = 6.8 Hz, 1H), 2.36 (s, 6H), 1.14 (d, J = 6.9 Hz, 6H).
13C NMR (CDCl3): δ 156.37, 142.49, 138.21, 137.57, 137.48, 136.38, 134.87, 128.95, 127.31, 127.27, 123.28, 107.78, 64.39, 64.12, 38.59, 30.01, 22.52, 21.41. Chloro[9-(3,5-dimethylphenyl)-7-isopropyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxin- 6-yl]dimethylsilane
nBuLi in hexanes (2.5 M, 5.0 ml, 12.5 mmol) was added in one portion to a solution of 5- (3,5-dimethylphenyl)-7-isopropyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxine (4.0 g, 12.5 mmol) in a mixture of 100 ml of ether and 10 ml of THF cooled to –50 °C. This mixture was stirred for 20 h at room temperature, then the resulting orange suspension was cooled to –50 °C, and dichlorodimethylsilane (7.5 ml, 7.95 g, 61.6 mmol, ca.5.0 equiv.) was added in one portion. The resulting mixture was stirred overnight at room temperature, then filtered through a glass frit (G4), and the filter cake was washed with 50 ml of hot toluene.
The combined filtrate was evaporated to dryness to give the title material which was used without further purification.
1H NMR (CDCl
3): δ 7.04 (br.s, 2H), 7.00 (s, 1H), 6.99 (s, 1H), 6.38 (br.s, 1H), 4.27-4.20 (m, 4H), 3.70 (s, 1H), 2.90-2.83 (m, 1H), 2.38 (s, 6H), 1.19 (d, J = 6.6 Hz, 3H), 1.11 (d, J = 6.9 Hz, 3H), 0.42 (s, 3H), 0.16 (s, 3H).
13C NMR (CDCl
3): δ 155.84, 140.39, 139.06, 137.84, 137.35, 135.86, 134.80, 128.83, 128.09, 123.04, 122.30, 111.85, 64.41, 64.13, 47.15, 29.36, 24.35, 21.46, 21.21, 1.07, -0.68. [9-(3,5-Dimethylphenyl)-7-isopropyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxin-6-yl] [9-(3,5-dimethylphenyl)-7-methyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxin-6-yl] dimethylsilane
nBuLi in hexanes (2.5 M, 5.0 ml, 12.5 mmol) was added in one portion to a solution of 5- (3,5-dimethylphenyl)-7-methyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxin (3.65 g, 12.5 mmol) in a mixture of 50 ml of ether and 50 ml of THF at –50 °C. This mixture was stirred overnight at room temperature, then the resulting yellow suspension was cooled to –50 °C, and 100 mg of CuCN was added. The obtained mixture was stirred for 15 min at –20 °C, and then a solution of ca.12.5 mmol of chloro[9-(3,5-dimethylphenyl)-7-isopropyl-2,3- dihydro-6H-indeno[5,6-b][1,4]dioxin-6-yl]dimethylsilane in 100 ml of ether was added in one portion. This mixture was stirred overnight at room temperature, then filtered through a pad of silica gel 60 (40-63 µm) which was additionally washed with 2x40 ml of ether. The combined yellowish elute was evaporated to dryness. The product was purified by flash chromatography on silica gel 60 (40-63 µm, 600 ml, eluent: hexanes:dichloromethane = 1:3, vol.). This procedure gave 7.10 g (84.9%, purity ca.97%) of the title product (as a ca 63:37 mixture of the stereoisomers) as a white solid. The product was used without further purification. Anti-dimethylsilanediyl[9-(3,5-dimethylphenyl)-7-isopropyl-2,3-dihydro-indeno[5,6- b][1,4]dioxin-6-yl][9-(3,5-dimethylphenyl)-7-methyl-2,3-dihydro-indeno[5,6-b][1,4] dioxin-6-yl] zirconium dichloride

BuLi in hexanes (2.5 M, 8.5 ml, 21.25 mmol) was added in one portion to a solution of [9- (3,5-dimethylphenyl)-7-isopropyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxin-6-yl][9-(3,5- dimethylphe-nyl)-7-methyl-2,3-dihydro-6H-indeno[5,6-b][1,4]dioxin-6-yl]dimethylsilane (7.10 g, 10.6 mmol) in 110 ml of di-n-butyl ether at room temperature. This mixture was stirred overnight at room temperature, then the resulting yellow suspension was cooled to 0 °C, and ZrCl4 (2.47 g, 10.6 mmol) was added. The reaction mixture was stirred for 24 h at room temperature to give an orange suspension. This suspension was evaporated to dryness. The formed solid was extracted with 50 ml of boiling toluene. On the evidence of NMR spectroscopy, the obtained extract included a ca.1:1 mixture of anti- and syn-MC- IE3 contaminated with polymeric by-products. This extract was evaporated to ca.30 ml, and 15 ml of hexane was added. The red solid precipitated from this solution after 15 min was collected and dried under vacuum. This procedure gave 2.2 g of syn-MC-IE3 as a solvate with ca.0.45 molecule of toluene and 0.2 molecule of hexane. Then 7 ml of hexane was added to the mother liquor. The orange solid precipitated from this solution overnight was collected and dried under vacuum, yielding 2.3 g of a ca.4:1 mixture of anti- and syn- isomers. Crystallization of this solid from a mixture of 10 ml of toluene and 3 ml of hexane gave 250 mg of a ca. 3:1 mixture of syn- and anti-isomers. The mother liquor was evaporated to ca.7 ml, and 5 ml of hexane was added. The orange solid precipitated from this solution at room temperature was collected and dried under vacuum. This procedure gave 1.2 g of a ca.93:7 mixture of anti- and syn-isomers. Recrystallization of this solid from a mixture of 5 ml of toluene and 3 ml of hexane gave 420 mg (4.8%) of pure anti-MC- IE3. Syn-MC-IE3: Anal. calc. for C . 44H46Cl2O4SiZr: C, 63.74; H, 5.59. Found: C, 63.98; H, 5.75.
1H NMR (CDCl
3): δ 7.18 (br.s, 4H), 7.05 (s, 1H), 7.03 (s, 1H), 6.96 (s, 1H), 6.94 (s, 1H), 6.47 (s, 1H), 6.43 (s, 1H), 4.23-4.16 (m, 4H), 4.11-4.05 (m, 4H), 3.03 (sep, J = 6.6 Hz, 1H), 2.34 (s, 6H), 2.33 (s, 6H), 2.24 (s, 3H), 1.36 (s, 3H), 1.33 (d, J = 6.6 Hz, 3H), 1.17 (s, 3H), 1.15 (d, J = 6.8 Hz, 3H).
13C NMR (CDCl
3): δ 148.02, 144.70, 144.65, 142.94, 142.76, 137.38, 137.35, 134.69, 133.09, 131.80, 131.30, 129.16, 127.69, 123.90, 123.15, 122.18, 121.50, 120.57, 112.99, 110.62, 110.30, 80.80, 79.62, 64.58, 64.10, 30.36, 29.50, 21.45, 21.41, 20.02, 17.99, 3.43, 2.69. Anti-MC-IE3: Anal. calc. for C
44H
46Cl
2O
4SiZr.: C, 63.74; H, 5.59. Found: C, 63.90 ; H, 5.76.
1H NMR (CDCl
3): δ 7.27 (br.s, 4H), 7.02 (s, 1H), 6.97 (s, 1H), 6.95 (s, 2H), 6.59 (s, 1H), 6.57 (s, 1H), 4.28-4.15 (m, 8H), 3.22 (sep, J = 6.7 Hz, 1H), 2.34 (s, 12H), 2.21 (s, 3H), 1.23 (s, 3H), 1.22 (s, 3H), 1.07 (d, J = 6.7 Hz, 3H), 1.05 (d, J = 6.7 Hz, 3H).
13C NMR (CDCl3): δ 147.65, 145.69, 145.18, 143.68, 143.15, 137.56 (br. s), 137.40, 135.09, 134.53, 134.50, 131.06, 129.28, 129.22, 128.60, 127.89, 127.79, 123.20, 123.07, 122.63, 119.92, 119.56, 114.87, 108.89, 108.08, 79.76, 77.87, 64.60, 64.56, 64.38, 64.23, 30.65, 29.19, 21.43, 21.37, 19.49, 18.87, 3.16, 2.77. Catalyst synthesis, used chemicals MAO Axion CA133030 wt-% solution in toluene was purchased from Chemtura/Lanxess and used as received and stored at –20 °C for not longer than 6 months. All the chemicals and chemical reactions were handled under an inert gas atmosphere using Schlenk and glovebox techniques, with oven-dried glassware, syringes, needles or cannulas. All catalysts have been prepared using silica Sunspera AGC DM-L-303, calcined at 600 °C. MAO Axion CA1330 was used as received and stored at –20 °C for not longer than 6 months. Catalyst preparations The catalysts were prepared by following a two-step preparation method. First step is the preparation of SiO2/MAO (activated carrier), followed by a second step where a toluene solution of the metallocene complex is impregnated on the dry support from the first step. Only in case the metallocene is not enough soluble in toluene (metallocenes MC-CE1 and
MC-CE2), a second aliquot of MAO is added to the metallocene/toluene slurry in order to promote the full dissolution of the metallocene. Preparation of SiO
2/MAO activated carrier A steel reactor equipped with a mechanical stirrer and a filter net was flushed with nitrogen. 10 kg of SiO2 carrier was first added from a feeding drum into the reactor, followed by careful pressurizing and depressurizing with nitrogen. Then, toluene (43.5 kg) was added. The SiO2/toluene slurry was stirred for 25 min at 22 °C. Then, 18 kg of 30 wt% MAO in toluene (Axion CA 1330) was added slowly (140 min) through a 12 mm line on the top of the reactor keeping the temperature around 22 °C. After MAO addition, the reactor temperature was quickly increased to 90 °C and the mixture was stirred at this temperature for 120 min. Then the hot toluene was filtered out and the solid cake was washed twice with hot toluene while stirring (43.5 kg, 90 °C, 30 min, 40 rpm). Each time the hot toluene was filtered out. Finally the solid cake was dried with slow stirring (5 rpm) under vacuum for 9 h at 80 °C. Synthesis of SiO2/MAO/MC-CE1 catalyst = Comparison catalyst 1 In a nitrogen filled glovebox, dry toluene (2.3 mL) and MAO (0.2 mL) were added to 20.8 mg of metallocene rac-dimethylsilandiyl[2-methyl-4-(4’-tert-butylphenyl)indenyl] [2-iso- propyl-4-(4’-tert-butylphenyl)indenyl]zirconium dichloride. The mixture was stirred for 30 minutes at room temperature. Next, 2.0 g of SiO2/MAO (prepared with PQ silica ES70X, synthesis SMLP013) was placed in a septum bottle. The solution of metallocene/MAO in toluene was added dropwise by means of a syringe to the SiO2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting powder was allowed to rest for 1 hour, then it was transferred into a Schlenk flask and dried under vacuum for 1 hour at 60 °C to yield 2.0 g of the catalyst as a free-flowing powder. Synthesis of SiO2/MAO/MC-CE2 catalyst = Comparison catalyst 2 In a nitrogen filled glovebox, dry toluene (2.5 mL) and MAO (0.1 mL) were added to 26.4 mg of metallocene rac-dimethylsilanediyl[2-methyl-4-(4’-tert-butylphenyl)-1,5,6,7- tetrahydro-s-indacen-1-yl])(2-isopropyl-4-(4’-tert-butyl-phenyl)indenyl) zirconium dichloride. The mixture was stirred for 30 minutes at room temperature. Next, 2.0 g of SiO2/MAO batch V397 was placed in a septum bottle. The solution of metallocene in toluene was added dropwise by means of a syringe to the SiO2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting powder was allowed to rest for 1 hour, then it was transferred into a Schlenk flask and dried under vacuum for 1 hour at 60 °C to yield 2.0 g of the catalyst as a light red free-flowing powder.
Synthesis of SiO2/MAO/MC-CE3 catalyst = Comparison catalyst 3 In a nitrogen filled glovebox, dry toluene (2.5 mL) was added to 27 mg of metallocene rac- dimethylsilanediylbis(2-methyl-4-(3’,5’-di-methyl phenyl)-5-methoxy-6-tert-butylinden-1-yl) zirconium dichloride. The solution was stirred for 30 minutes at room temperature. Next, 2.0 g of SiO
2/MAO batch V407 was placed in a septum bottle. The solution of metallocene in toluene was added dropwise by means of a syringe to the SiO
2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting powder was allowed to rest for 1 hour, then it was transferred into a Schlenk flask and dried under vacuum for 1 hour at 60 °C to yield 2.0 g of the catalyst as a red free-flowing powder. Synthesis of SiO2/MAO/MC-CE4 catalyst = Comparison catalyst 4 In a nitrogen filled glovebox, dry toluene (2.5 mL) was added to 29.9 mg of metallocene rac-dimethylsilanediylbis(2-neopentyl-4-(3’,5’-dimethylphenyl)-5-methoxy-6-tert- butylinden-1-yl) zirconium dichloride. The solution was stirred for 30 minutes at room temperature. Next, 2.0 g of SiO2/MAO was placed in a septum bottle. The solution of metallocene in toluene was added dropwise by means of a syringe to the SiO2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting powder was allowed to rest for 1 hour, then it was transferred into a Schlenk flask and dried under vacuum for 1 hour at 60 °C to yield 2.0 g of the catalyst as a pink free-flowing powder. Synthesis of SiO2/MAO/ MC-IE1 catalyst = Inventive catalyst 1 (IE1) In a nitrogen filled glovebox, dry toluene (2.5 mL) was added to 29.6 mg of metallocene rac-dimethylsilanediyl[2-methyl-4-(3’,5’-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl] [2-isopropyl-4-(3’,5’-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl] zirconium dichloride. The mixture was stirred for 30 minutes at room temperature. Next, 2.0 g of SiO2/MAO was placed in a septum bottle. The solution of metallocene in toluene was added dropwise by means of a syringe to the SiO2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting powder was allowed to rest for 1 hour, then it was transferred into a Schlenk flask and dried under vacuum for 1 hour at 60 °C to yield 2.0 g of the catalyst as a light salmon-red free-flowing powder. Synthesis of SiO2/MAO/MC-IE2 catalyst = Inventive catalyst 2 (IE2) In a nitrogen filled glovebox, dry toluene (2.5 mL) was added to 30.1 mg of metallocene rac-dimethylsilanediyl[2-methyl-4-(3’,5’-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl] [2-isopropyl-4-(3’,5’-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl] hafnium dichloride. The mixture was stirred for 30 minutes at room temperature. Next, 2.0 g of of SiO
2/MAO was placed in a septum bottle. The solution of metallocene in toluene was added dropwise
by means of a syringe to the SiO
2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting powder was allowed to rest for 1 hour, then it was transferred into a Schlenk flask and dried under vacuum for 1 hour at 60 °C to yield 2.0 g of the catalyst as a yellow free-flowing powder. Synthesis of SiO2/MAO/MC-IE3 catalyst = Inventive catalyst 3 In a nitrogen filled glovebox, dry toluene (2.5 mL) was added to 25.0 mg of metallocene IE3. The mixture was stirred for 30 minutes at room temperature. Next, 2.0 g of the silica/MAO carrier was placed in a glass vial. The solution of metallocene in toluene was added dropwise by means of a syringe to the SiO
2/MAO carrier over the course of 5 minutes with gentle mixing. The resulting mixture was shaken well and allowed to stay for 1 hour. The resulting solid was dried under vacuum for 1 hour at 60°C to yield the catalyst as a beige free-flowing powder. The metallocene content in each catalyst is calculated by mass balance. The values are listed in Table 1: Table 1: Catalysts tested and their metallocene content MC in Catalyst MC Al catalyst * type wt% wt% CE1 MC-CE1 13.6 1.00 CE2 MC-CE2 13.2 1.28 CE3 MC-CE3 13.2 1.33 CE4 MC-CE4 13.1 1.48 IE1 MC-IE1 13.8 1.33 IE2 MC-IE2 13.8 1.48 IE3 MC-IE3 12.4 1.23 * metallocene content in the dry catalyst calculated from mass balance
Polymerization examples Reference examples 1-12 and 22-25 and Inventive Examples 13-21 and 26-30: Preparation of polypropylene homopolymers Propylene polymerisation procedure (liquid propylene, 5-L reactor) A stainless-steel reactor equipped with a ribbon stirrer, with a total volume of 5.3 dm³ was filled with 800 g propylene. Triethylaluminium (0.3 ml of 0.62 molar solution in n-heptane) was added using a stream of 150 g propylene, then the chosen amount of H2 (see Tables) was added via mass flow controller in one minute. The reactor temperature was stabilized at 20 °C (HB-Therm) and the solution was stirred and 250 rpm for at least 20 min. Then the catalyst was injected as described in the following. The desired amount of solid catalyst was loaded into a 5 ml stainless steel vial and then flushed into the reactor with 150 g propane. Stirring speed was kept at 250 rpm and pre- polymerisation was run for 10 minutes at 25 °C. Then the polymerisation temperature was increased to the set value. The second aliquot of H2 was added at 30 °C over 6 min. The reactor temperature was kept constant throughout the polymerisation. The polymerisation time was measured starting when the temperature was 2 °C below the set polymerisation temperature. When the polymerisation time of 60 min has lapsed, the reaction was stopped by injecting 5 ml ethanol, cooling the reactor and simultaneously flashing the volatile components. After purging the reactor 3 times with N2 and one vacuum/N2 cycle, the reactor was opened, the polymer powder was taken out and dried overnight in a fume hood.100 g of the polymer was additivated with 0.5 wt% Irganox B225 (dissolved in acetone) and then dried overnight in a fume hood and additionally one hour in a vacuum drying oven at 60 °C. Propylene polymerisation procedure (liquid propylene, 20-L reactor) A stainless-steel reactor equipped with a ribbon stirrer, with a total volume of 20.9 dm³ containing 0.2 bar-g propylene, was filled with 3.95 kg propylene. Triethylaluminium (0.8 ml of 0.62 molar solution in n-heptane) was added using a stream of 250 g propylene, then 0.5 NL of H2 was added via mass flow controller in one minute. The reactor temperature was stabilized at 25 °C (HB-Therm) and the solution was stirred and 250 rpm for at least 20 min. Then the catalyst was injected as described in the following. The desired amount of solid catalyst was loaded into a 5 ml stainless steel vial. This feeder system was mounted on a port on the lid of the autoclave. Then the catalyst was flushed into the reactor with 350 g propylene. Stirring speed was kept at 250 rpm and pre- polymerisation was run for 10 minutes at 25 °C or 30 °C. Then the polymerisation
temperature was increased to 75 °C. The second aliquot of H2 was added at 30 °C over 1 min. The reactor temperature was kept constant throughout the polymerization. The polymerization time was measured starting when the temperature was 2 °C below the set polymerization temperature. When the set polymerization time has lapsed, the reaction was stopped by injecting 5 ml ethanol, cooling the reactor and simultaneously flashing the volatile components. After purging the reactor 3 times with N2 and one vacuum/N2 cycle, the reactor was opened, the polymer powder was taken out and dried overnight in a fume hood.100 g of the polymer was additivated with 0.5 wt% Irganox B225 (dissolved in acetone) and then dried overnight in a fume hood and additionally one hour in a vacuum drying oven at 60 °C. Propylene polymerisation results We have compared the performance of the catalysts prepared from the seven different metallocenes MC-CE1, MC-CE2, MC-CE3, MC-CE4, and MC-IE1, MC-IE2, MC-IE3 in liquid propylene polymerisation. The conditions are described in Table 2A and the results are shown in Table 2B: examples 1-21 have been produced in the 5-L reactor, while examples 22-29 have been produced in the 20-L reactor. The hPP melting points obtained with the seven catalysts are compared in Figure 1. Table 2A: Propylene polymerisation conditions tnu
e r 2 t o
e r o n n Hn n
e r l ep s l
u y m
t f l a
a r e e
y i bo d
i o i t f i u t og
i o i t a r e ma
a x
t t e m a s p
i t d p e e
e r f s 2n
.p
i n a d s en e a p m
i t e c
l y a t m
f p
r t m
r t m a
e t 2 H e T
e f e T c H mg °C min NL NL °C °C min 1 CE1 16.5 25 10 0.13 0.63 30 75 60 2 CE1 18.1 25 10 0.13 0.38 30 75 60 3 CE1 19.5 25 10 0.13 0.13 30 75 60 4 CE2 23 25 10 0.13 0.63 30 75 60 5 CE2 29 25 10 0.13 0.13 30 75 60 6 CE2 23 25 10 0.13 0.88 30 75 60 7 CE3 17 25 10 0.13 0.63 30 75 60 8 CE3 17 25 10 0.13 1.37 30 75 60 9 CE3 23 25 10 0.13 0.13 30 75 60 10 CE4 14 25 10 0.13 0.63 30 75 60 11 CE4 11 25 10 0.13 1.38 30 75 60 12 CE4 25 25 10 0.13 0.13 30 75 60 13 IE1 15 25 10 0.13 0.63 30 75 60 14 IE1 21 25 10 0.13 0.63 30 75 60
15 IE1 17 25 10 0.13 1.37 30 75 60 16 IE1 25 25 10 0.13 0.12 30 75 60 17 IE1 17 30 10 3.70 0.00 30 75 60 18 IE1 17 30 10 0.75 0.00 30 75 60 19 IE1 17 30 10 1.50 0.00 30 75 60 20 IE1 26 30 10 0.25 0.00 30 75 60 21 IE2 34 25 10 0.13 0.63 30 75 60 22 CE2 81 30 10 0.5 0.5 30 70 60 2
3 CE3 64 30 10 0.50 1.50 30 75 60 24 CE3 56 30 10 0.50 2.00 30 75 60 2
5 CE3 70 30 10 0.50 1.00 30 75 60 26
IE1 65 30 10 0.50 1.00 30 75 60 2
7 IE1 68 30 10 0.50 1.50 30 75 60 28 IE1 67 30 10 0.50 2.00 30 75 60 2
9 IE3 109 30 10 0.50 1.01 30 75 60 Table 2B: Propylene polymerisation results y y
i i t f re re t v
i t oy d
f d l
e ts v c
i t w
ow n p m
l y l d i tc u s o d n p o r 2p
r Tm
M / a
a t i e u
o e R Mw w mrrm 2.1e 3.1 x a e c Y d o
r r p d e F e P C
l km M u
l y Mm l
y M Bo p o p g kg/gcat kg/gM C g/ml g/10min °C g/mol mol% mol% mol% 1 CE1 99 6.0 527 0.48 208 154.1 98100 2.3 n/a n/a n/a 2 CE1 91 5.0 442 0.5 47.8 155 137000 2.4 n/a n/a n/a 3 CE1 53 2.7 237 0.48 7.5 155 216000 2.6 n/a n/a n/a 4 CE2 213 9.3 724 0.48 61.5 157.3 131500 2.3 0.15 0.24 0.19 5 CE2 118 4.1 318 0.49 1.6 156.9 296000 2.4 n/a n/a n/a 6 CE2 203 8.8 690 0.47 1210 154.8 60850 2.6 n/a n/a n/a 7 CE3 201 11.8 889 0.5 0.6 152.4 478000 2.9 n/a n/a n/a 8 CE3 313 18.4 1384 0.46 22.8 150.8 184500 3.5 n/a n/a n/a 9 CE3 185 8.0 605 0.49 0.0 150.651320000 2.6 n/a n/a n/a 10 CE4 276 19.4 1312 0.5 2.9 152 307500 2.6 n/a n/a n/a 11 CE4 215 19.5 1321 0.43 223 151.3 99200 3.0 n/a n/a n/a 12 CE4 328 13.1 886 0.49 0.0 150.91045000 3.0 n/a n/a n/a 13 IE1 155 10.3 777 0.49 13.6 160.8 186000 2.3 0.05 0.22 0.06 14 IE1 205 9.8 734 0.49 10.5 160.4 200000 2.2 n/a n/a n/a 15 IE1 185 10.9 818 0.45 689 158.4 68500 2.4 n/a n/a n/a 16 IE1 62 2.5 186 0.48 0.3 160.6 505000 2.8 n/a n/a n/a 17 IE1 262 15.4 1159 0.44 1894 157.7 49350 2.9 n/a n/a n/a 18 IE1 213 12.5 942 0.46 79.1 159.8 123500 2.3 n/a n/a n/a 19 IE1 332 19.5 1468 0.46 665 158.5 68650 2.3 n/a n/a n/a 20 IE1 138 5.3 399 0.49 1.0 160.8 364000 2.5 n/a n/a n/a 21 IE2 20 0.6 40 0.47 6.6 162.7 224000 3.6 0.08 0.06 0.10
22 CE2 723 8.9 696 0.47 20.2 157 174000 2.4 0.10 0.29 0.12
23 CE3 1078 16.84 1266 0.48 1.0 150.7 402000 3.4 n/a n/a n/a 24 CE3 1026 18.32 1378 0.48 3.1 150.9 312500 3.4 0 1,01 0
25 CE3 1065 15.21 1144 0.48 0.3 150 551000 3.3 n/a n/a n/a 26
IE1 625 9.615 718 0.44 10.2 161.1 207000 2.5 0 0,23 0,04
27 IE1 848 12.47 931 0.44 30.0 160.3 151500 2.4 n/a n/a n/a 28 IE1 888 13.25 989 0.44 78.0 160.4 119500 2.5 n/a n/a n/a
29 IE3 705 6.468 524 0.41 220 157.5 90150 2.3 n/a n/a n/a Note how the effect of different substituents is not additive: in terms of hPP melting point MC-IE1 performs much better than the two comparison catalysts based on MC-CE1 and MC-CE2, making this finding unexpected It is also important to note that substituents larger than methyl but without alpha substituents (e.g. neopentyl in MC-CE4) do not increase hPP Tm, therefore (at least) one alpha-branched 2-substituent is necessary to achieve this high melting point. In addition, MC-IE1 has a better molecular weight / activity balance than both MC-CE1 and MC-CE2, as shown in Figure 2. Reference examples 30 and Inventive Examples 31-34: Preparation of heterophasic propylene copolymers Polymerization procedure Monomers and gases Hydrogen (quality 6.0) was supplied by Air Liquide and used as received. Propylene, quality 2.3, and ethylene have been purified by passing through columns filled with PolyMax301 T-4427B (60°C; Cu/CuO), Molecular sieve MS13X-APG 1/16 and Selexsorb COS 1/8. Propylene and propylene/ethylene 2-step polymerization procedure (20-L reactor, liquid + gas phase) Step 1: Propylene homo polymerization in bulk, 20-L reactor A stainless-steel reactor equipped with a ribbon stirrer, with a total volume of 20.9 dm³ containing 0.2 bar-g propylene, was filled with additional 4.45 kg propylene. Triethylaluminium (0.8 ml of 0.62 molar solution in n-heptane) was added using a stream of 250 g propylene, then the chosen amount of H2 (see Tables) was added via mass flow controller in one minute. The reactor temperature was stabilized at 25 °C (HB-Therm) and the solution was stirred and 250 rpm for at least 20 min.
Then the catalyst was injected as described in the following. The desired amount of solid catalyst was loaded into a 5 ml stainless steel vial; alternatively the chosen amount of a catalyst slurry in oil, after shaking for about 5 min in a glass vial, was drawn with a syringe and loaded into the 5 ml stainless steel vial. Then the catalyst vial was mounted on a port on the lid of the reactor. The catalyst was fed into the reactor by flushing 350 g propylene from the balance through the catalyst vial. Stirring speed was kept at 250 rpm and pre-polymerization was run for 10 minutes at either 25 or 30 °C. Then the polymerization temperature was increased to the set value. The second aliquot of H2 was added at either 30 or 60 °C over 2 min. The reactor temperature was kept constant throughout the polymerization. The polymerization time was measured starting when the temperature was 2 °C below the set polymerization temperature. When the set polymerization time has lapsed, the reaction was stopped by injecting 5 ml ethanol, cooling the reactor and simultaneously flashing the volatile components. After purging the reactor 3 times with N2 and one vacuum/N2 cycle, the reactor was opened, the polymer powder was taken out and dried overnight in a fume hood. 100 g of the polymer was additivated with 0.5 wt% Irganox B225 (dissolved in acetone) and then dried overnight in a fume hood and additionally one hour in a vacuum drying oven at 60 °C. Step 2: Ethylene-propylene copolymerization in gas phase, 20-L reactor Step 2 was performed as follows. After the bulk homopolymerization step was completed, the stirrer speed was reduced to 50 rpm and the pressure was reduced to 0.4 bar-g by venting the monomers. The stirrer speed was set to 180 rpm and the reactor temperature was set to 70 °C. Then the reactor pressure was increased to 20 bar-g by feeding a defined C3/C2 gas mixture (see tables). The C3/C2 ratio was defined by
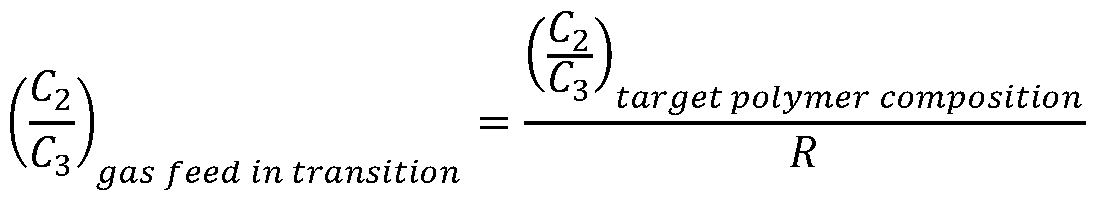
where C2/C3 was the weight ratio of the two monomers and R was their relative reactivity ratio, determined experimentally. In the present experiments, the value of R was set at 0.40. The temperature was held constant by thermostat and the pressure was kept constant by feeding via mass flow controller a C3/C2 gas mixture of composition corresponding to the target polymer composition and by thermostat, until the set time for this step had expired. Then the reactor was cooled down (to about 30 °C) and the volatile components flashed out. After purging the reactor 3 times with N2 and one vacuum/N2 cycle, the product was
taken out and dried overnight in a fume hood.100 g of the polymer was additivated with 0.5 wt% Irganox B225 (solution in acetone) and dried overnight in a hood followed by 2 hours in a vacuum drying oven at 60°C Heterophasic polymerization results The performance of the catalysts prepared from three different metallocenes MC-CE2, MC-IE1 and MC-IE3 in liquid propylene polymerization. The propylene polymerization conditions are shown in Table 3A. The results are shown in Table 3B. Table 3A: Polymerization conditions Prepoly step Transition tnuo . g n p
i n i o o i
t t l y l
e t p s m . a p e mde sn op
l m
l y t m H2 H2 e
t e
a a
a r t s
e k u x a
l y e
i m T T
t r f a t 2
t r H e
p b E C
a t a S
f o
i mm C To
r f mg °C min NL NL °C min 30 CE2 89 25 10 0,5 2,5 60 19 31 CE3 57 25 10 0,5 2,5 60 18 32 IE1 109 30 10 0,5 1,0 30 16 33 IE3 127 30 10 0,5 1,0 30 16 (Table 3A continued.) Bulk step Gas phase step n s s l
e i o a a s s p .
i t g g a a p e s o n
i n n
i n
e r n .
g g m n
t es d
i o i t d
i o i t u
i e s 2 s p e n
i es n
i es a m x e
i m a T
r t l k u a e h
f s e n
f s s
Hah me
i m d ah d ah E T e
b p 2 3 n
e Ca
r t Ca
r t r d p T e p e p P e T
f f i
mm To d 2 3 r
f d C C A °C min min g g barg NL °C min g g 30 75 40 11 250,3 304,8 20 0 70 120157,6 473,0 31 75 40 11 239,4 312,1 20 0 70 120154,3 464,6 32 75 24 11 266,7 277,1 20 0,1 70120 20,9 55,3 33 75 24 11 261,6 280,4 20 0,1 70 40 59,7 178,4 Table 3B: Results on whole material Analysis whole material
y l
e l l i t e y l
l n
i t y
i t y v y
i t es e v a n
i s p m
l d i
a r v
i t e c
a r ec v
i t i t l k e o c c u
i tv
l k i t u
i tc h p y
i t ah a
e x Y v u v
l u u
b b u s v
i p E Od o
r O
a t d d e p
o r o r i n c A
i n d o
r a
t g c sa m p P P
i n A g g kg/g
cat kg/g
MC kg
PP/g
cat kg
PP/g
cat/h kgPP/g
cat kgPP/g
cat/h 30 1140 12,8 1001 6,0 9,0 6,8 3,1 31 1195 21,0 1580 10,5 15,8 10,4 4,8 32 510 4,7 350 3,5 8,8 0,5 0,2 33 664 5,2 424 3,4 8,5 1,8 2,1 (Table 3B continued.) c n o
i r y e e y
i t 2 re l
e l e
i t sa s d
l k s Rd p
a t n a m e e v
i t g h wu n F w T
c H
c T
m H
m p o b e Mo a M c x a P d p E kgPP/gMC/h g/ml g/10min °C J/g °C J/g 30 244 0,38 62,8 116 51 157 52 31 360 0,47 1,1 113,2 47 150,4 47 32 16 0,46 7,4 115 97 160 100 33 173 0,42 22 116 72 159 73 Table 3C: Results on soluble fraction l
e Analysis soluble fraction p m Crystex GPC ax SF iV (SF) C2 (SF) RC2/C3 Mw Mw/Mn E wt% dl/g wt% g/mol 30 54,3 3,4 23,19 0,37 338000 3,7 31 50 3,7 21,8 0,36 456000 3,1 32 7,9 4,6 24,5 0,34 608000 2,4 33 32,8 4,5 30,4 0,47 505000 3,1 e
l (Table 3C continued.) p m Solution 13C NMR ax E C2 C2 R
C2/C3 2,1e E-2,1 r
Exr
P wt% mol% mol% mol% 30 23,4 31,43 0,37 0 0,18 1,5 31 21,4 28,9 0,35 0 0,14 1,4 32 25,2 33,6 0,35 0 0 1,6 33 31,6 40,9 0,50 0 0 0,9 Rubber iV is higher for IE1 and IE3 and the matrix (hPP) melting point is also higher for IE1 and IE3 compared to CE2 and CE3.
Ethylene reactivity of IE3 (R
C2/C3~0.5) is significantly higher than that of both CE2 and CE3 (R
C2/C3~0.35 - 0.37).
 Racemic Anti Racemic Syn Preferred metallocene catalyst complexes are in the anti-configuration. The metallocene complexes of the invention are preferably employed as the racemic-anti- isomers. Ideally, therefore at least 95 mol%, such as at least 98 mol%, especially at least 99 mol% of the metallocene catalyst complex is in the racemic anti-isomeric form. For the purpose of this invention, the numbering scheme of the indenyl ligands is the following:
Racemic Anti Racemic Syn Preferred metallocene catalyst complexes are in the anti-configuration. The metallocene complexes of the invention are preferably employed as the racemic-anti- isomers. Ideally, therefore at least 95 mol%, such as at least 98 mol%, especially at least 99 mol% of the metallocene catalyst complex is in the racemic anti-isomeric form. For the purpose of this invention, the numbering scheme of the indenyl ligands is the following:
 The present metallocene catalyst complexes require the combination of two distinctive features of the ligand framework: 1: specific 5, 6 substitution of the indenyl ligands, and 2: one alpha-branched substituent on the 2-position of one of the indenyl ligands. The present invention accordingly relates to metallocene complexes of formula (I)
The present metallocene catalyst complexes require the combination of two distinctive features of the ligand framework: 1: specific 5, 6 substitution of the indenyl ligands, and 2: one alpha-branched substituent on the 2-position of one of the indenyl ligands. The present invention accordingly relates to metallocene complexes of formula (I)
 wherein Mt is Zr or Hf, preferably Zr; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl;
R2’ is alpha-branched C3-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; n are each independently an integer from 1 to 5; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; L is O or S; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or -OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent LR51 and R6 and/or LR51’ and R6’ form together a -O[C(R62)2]mO- group, with R62 being each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl.
Preferably R2 is CH2-R21, with R21 being H, linear C1-C6-alkyl, such as methyl, ethyl, n- propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl. Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. It is preferred that each n is independently, same or different from each other, an integer from 1 to 3, such as 1, 2 or 3. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3´-, 4´-, and/or 5´-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the two phenyl rings have different substitution patterns. It is therefore preferred when n is 1, the only R3 and/or R4 group, respectively, is preferably in the para position. If n is 2, then the two R3 and/or R4 groups, respectively are preferably in the meta positions. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C10-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. As noted above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl.
Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3 or –OR61, with R61being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R62)2]mO- group, each R62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-a)
wherein Mt is Zr or Hf, preferably Zr; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl;
R2’ is alpha-branched C3-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; n are each independently an integer from 1 to 5; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; L is O or S; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or -OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent LR51 and R6 and/or LR51’ and R6’ form together a -O[C(R62)2]mO- group, with R62 being each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl.
Preferably R2 is CH2-R21, with R21 being H, linear C1-C6-alkyl, such as methyl, ethyl, n- propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl. Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. It is preferred that each n is independently, same or different from each other, an integer from 1 to 3, such as 1, 2 or 3. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3´-, 4´-, and/or 5´-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the two phenyl rings have different substitution patterns. It is therefore preferred when n is 1, the only R3 and/or R4 group, respectively, is preferably in the para position. If n is 2, then the two R3 and/or R4 groups, respectively are preferably in the meta positions. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C10-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. As noted above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl.
Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3 or –OR61, with R61being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R62)2]mO- group, each R62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-a)
 wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not H; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and
R6 and R6’ are each independently, same or different from each other, C(R61)3 or -OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR51 and R6 and/or OR51’ and R6’ form together a –O[C(R62)2]mO- group, with R62 being each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I-a), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-a) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl. Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each
other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3´-, 4´-, and/or 5´-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. It is therefore preferred if one or two R3 and/or R4 groups is H. If two R3 and/or R4 groups are H then the remaining R3 and/or R4 group, respectively, is preferably in the para position. If one R3 and/or R4 group is H then the remaining R3 and/or R4 groups are preferably in the meta positions. Advantageously one or two R3 on the phenyl group are not H, when two R3 are not H preferably these two R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the indenyl moiety preferably one or two R4 on the phenyl group are not H, more preferably two R4 are not H, and most preferably these two R4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. As mentioned above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl. Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3 or –OR61, with R61 being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R62)2]mO- substituent, each R62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2.
Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-b)
wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not H; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and
R6 and R6’ are each independently, same or different from each other, C(R61)3 or -OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR51 and R6 and/or OR51’ and R6’ form together a –O[C(R62)2]mO- group, with R62 being each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I-a), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-a) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl. Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each
other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3´-, 4´-, and/or 5´-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. It is therefore preferred if one or two R3 and/or R4 groups is H. If two R3 and/or R4 groups are H then the remaining R3 and/or R4 group, respectively, is preferably in the para position. If one R3 and/or R4 group is H then the remaining R3 and/or R4 groups are preferably in the meta positions. Advantageously one or two R3 on the phenyl group are not H, when two R3 are not H preferably these two R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the indenyl moiety preferably one or two R4 on the phenyl group are not H, more preferably two R4 are not H, and most preferably these two R4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. As mentioned above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl. Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3 or –OR61, with R61 being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R62)2]mO- substituent, each R62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2.
Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-b)
 (I-b) wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R3 and R4 are each independently H, C1-C10-hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or -OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR51 and R6 and/or OR51’ and R6’ form together a -O[C(R62)2]mO- group, wherein R62 are each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I-b), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-b) it is preferred if Mt is Zr or Hf, preferably Zr.
Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably H, linear or branched C1-C4-alkyl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. It is therefore preferred if one or two R3 and/or R4 groups is H. If two R3 and/or R4 groups are H then the remaining R3 and/or R4 group, respectively, is preferably in the para position. If one R3 and/or R4 group is H then the remaining R3 and/or R4 groups are preferably in the meta positions. Advantageously one or two R3 on the phenyl group are not H, more preferably R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl.
For the indenyl moiety preferably one or two R4 on the phenyl group are not H, more preferably two R4 are not H, and most preferably these two R4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. As mentioned above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl. Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3 or –OR61, with R61 being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R62)2]mO- substituent, each R62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (l-c)
(I-b) wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R3 and R4 are each independently H, C1-C10-hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or -OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR51 and R6 and/or OR51’ and R6’ form together a -O[C(R62)2]mO- group, wherein R62 are each independently, same or different from each other, H or linear or branched C1- C6-alkyl, with m being 1 to 3. For the above-defined metallocene complexes of formula (I-b), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-b) it is preferred if Mt is Zr or Hf, preferably Zr.
Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably H, linear or branched C1-C4-alkyl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.4´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. It is therefore preferred if one or two R3 and/or R4 groups is H. If two R3 and/or R4 groups are H then the remaining R3 and/or R4 group, respectively, is preferably in the para position. If one R3 and/or R4 group is H then the remaining R3 and/or R4 groups are preferably in the meta positions. Advantageously one or two R3 on the phenyl group are not H, more preferably R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl.
For the indenyl moiety preferably one or two R4 on the phenyl group are not H, more preferably two R4 are not H, and most preferably these two R4 are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. As mentioned above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl. Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3 or –OR61, with R61 being each independently, same of different from each other, linear C1- C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl or OMe, most preferably tert-butyl. Advantageously in the -O[C(R62)2]mO- substituent, each R62 is independently H or methyl, preferably H, and m is 1 to 3, such as 1, 2 or 3, preferably 2. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (l-c)
 (I-c) wherein Mt is Zr or Hf; X is a sigma ligand
R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, C1-C10-hydrocarbyl group, -OR31, -SR31, or -N(R31)2, with R31 being C1-C10-hydrocarbyl; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3, with R61 being each independently, same of different from each other, linear or branched C1-C6- alkyl. For the above-defined metallocene complexes of formula (I-c), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-c) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl.
Preferably R2’ is CH(R21’)2 or SiH(R21’)2, wherein R21’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably linear or branched C1-C4-alkyl or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially methyl, or tert-butyl. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.3´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously one or two R3 on the phenyl group are not H, more preferably R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the second indenyl moiety preferably R4 are the same like 3´,5´-di-methyl or 3´,5´-di- tert-butyl. As mentioned above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl. Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3, wherein R61 are each independently, same of different from each other, linear C1-C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl.
Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-d)
(I-c) wherein Mt is Zr or Hf; X is a sigma ligand
R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, C1-C10-hydrocarbyl group, -OR31, -SR31, or -N(R31)2, with R31 being C1-C10-hydrocarbyl; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3, with R61 being each independently, same of different from each other, linear or branched C1-C6- alkyl. For the above-defined metallocene complexes of formula (I-c), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-c) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl. Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl.
Preferably R2’ is CH(R21’)2 or SiH(R21’)2, wherein R21’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably linear or branched C1-C4-alkyl or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially methyl, or tert-butyl. Preferably, the R3 and R4 substituents of the respective phenyl ring are in the 3-, 4-, and/or 5-position of the ring, whereby the 1-position is attached to the indenyl ring. It is for example possible that the phenyl ring substituted in the para position i.e.3´ position only, like 4´-tert.-butyl phenyl, or di-substituted in the meta positions, i.e.3´ and 5´ position, like 3´,5´-dimethylphenyl or 3´,5´-ditert.-butylphenyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously one or two R3 on the phenyl group are not H, more preferably R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the second indenyl moiety preferably R4 are the same like 3´,5´-di-methyl or 3´,5´-di- tert-butyl. As mentioned above, R51’ is C1-C10-hydrocarbyl, for example, a linear or branched C1-C10- hydrocarbyl. Preferably R51 and R51’ are each independently, same of different from each other, linear or branched C1-C6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, sec-butyl, or tert-butyl, C7-C10-arylalkyl, C7-C10-alkylaryl, or C6-C10-aryl, more preferably linear C1-C6-alkyl, branched C3-C6-alkyl, or C6-aryl, even more preferably linear C1-C4-alkyl, yet even more preferably methyl or ethyl, and most preferably methyl. It is preferred that R51 and R51’ are the same. Most preferably, both R51 and R51’ are methyl. Preferably R6 and R6’ are each independently, same or different from each other, C(R61)3, wherein R61 are each independently, same of different from each other, linear C1-C3-alkyl; more preferably methyl. It is preferred that R6 and R6’ are the same. Advantageously, both R6 and R6’ are tert-butyl.
Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-d)
 wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, C1-C10-hydrocarbyl group, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl. For the above-defined metallocene complexes of formula (I-d), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-d) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl.
Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl. Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably linear or branched C1-C4-alkyl; even more preferably, each R3 and R4 are each independently, same or different from each other, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially methyl, or tert-butyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously R3 are the same, like 3´,5´-di-methyl. For the indenyl moiety preferably two R4 of each phenyl ring are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-e)
wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-, preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, C1-C10-hydrocarbyl group, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl. For the above-defined metallocene complexes of formula (I-d), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-d) it is preferred if Mt is Zr or Hf, preferably Zr. Each X is a sigma ligand. Preferably, each X is independently, same or different from each other, H, halogen, C1-C6-alkoxy, or R´ group, where R´ is C1-C6-alkyl, phenyl, or benzyl. More preferably, each X is independently, same or different from each other, Cl, benzyl, or methyl. It is preferred that both X groups are the same. Most preferably both X are Cl, methyl, or benzyl, especially Cl.
Preferably R1 are each independently, same or different from each other, C1-C10- hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10-cycloalkyl-alkyl, C7- C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert- butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl. Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably linear or branched C1-C4-alkyl; even more preferably, each R3 and R4 are each independently, same or different from each other, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially methyl, or tert-butyl. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously R3 are the same, like 3´,5´-di-methyl. For the indenyl moiety preferably two R4 of each phenyl ring are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. Viewed from another aspect the invention provides a metallocene catalyst complex of formula (I-e)
 wherein R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form, together with the Si atom they are attached to, a C4-C8-ring; R2 is linear C1-C20- preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not H. For the above-defined metallocene complexes of formula (I-e), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-e) R1 are preferably each independently, same or different from each other, C1-C10-hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10- cycloalkyl-alkyl, C7-C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert-butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl.
Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably H, linear or branched C1-C4-alkyl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the indenyl moiety preferably two R4 of each phenyl ring are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. For the avoidance of doubt, any narrower definition of a substituent offered above can be combined with any other broad or narrowed definition of any other substituent. Throughout the disclosure above, where a narrower definition of a substituent is presented, that narrower definition is deemed disclosed in conjunction with all broader and narrower definitions of other substituents in the application. Intermediates Whilst the invention primarily relates to catalysts complexes, it will be appreciated that the ligands used to form those complexes are also new. The novel indenes of the present invention bear the combination of the distinctive features of the metallocene ligand framework: 1: 5-alkoxy indene, preferably methoxy indene, with 6-tertiary hydrocarbyl, preferably tertiary alkyl, substituent, and 2: an alpha-branched alkyl substituent on the 2-position of the indene.
The present invention accordingly further relates to indenes of formula (II)
wherein R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form, together with the Si atom they are attached to, a C4-C8-ring; R2 is linear C1-C20- preferably C1-C10-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not H. For the above-defined metallocene complexes of formula (I-e), the following represent preferable embodiments, which can be selected alone or in combination: In a complex of formula (I-e) R1 are preferably each independently, same or different from each other, C1-C10-hydrocarbyl, more preferably C1-C10-alkyl, C4-C10-cycloalkyl, C5-C10- cycloalkyl-alkyl, C7-C10-arylalkyl, C6-C10-aryl, or C7-C10-alkylaryl, such as methyl, ethyl, propyl, isopropyl, tert-butyl, isobutyl, C3-C8-cycloalkyl, cyclohexylmethyl, phenyl, or benzyl, even more preferably both are C1-C6-alkyl, C5-C6-cycloalkyl, or C6-aryl. In an embodiment each R1 is independently, same or different from each other, C1-C10-alkyl, optionally substituted with C1-C10-alkoxy. It is preferred that both R1 groups are the same. Most preferably, both R1 are methyl. Preferably R2 is CH2-R21, with R21 being H, linear C1-6-alkyl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, preferably R21 being H or linear C1-C3-alkyl; more preferably R2 is methyl or ethyl, most preferably methyl.
Preferably R2’ is CH(R21’)2 or SiH(R21’)2, with R21’ being each independently, same of different from each other, linear or branched C1-C6-alkyl, C3-C8-cycloalkyl, or C6-C9-aryl, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, tert-butyl, cyclohexyl, or phenyl, more preferably R21’ being linear or branched C1-C6-alkyl; more preferably, R2’ is CH(CH3)2 or SiHMe2, even more preferably CH(CH3)2. Preferably R3 and R4 are each independently, same or different from each other, H, linear or branched C1-C6-alkyl, C6-C20-aryl, or -OR31, with R31 being C1-C4-hydrocarbyl; more preferably H, linear or branched C1-C4-alkyl, or -OR31, with R31 being C1-C4-hydrocarbyl; even more preferably, each R3 and R4 are each independently, same or different from each other, H, methyl, ethyl, isopropyl, tert-butyl, or methoxy, especially H, methyl, or tert-butyl, whereby at least one R3 and at least one R4 is not H. Furthermore, it is possible that each of the phenyl rings have the same substitution pattern or that the three phenyl rings have different substitution patterns. Advantageously R3 are the same, like 3´,5´-di-methyl or 4´- tert-butyl. For the indenyl moiety preferably two R4 of each phenyl ring are the same like 3´,5´-di- methyl or 3´,5´-di-tert-butyl. For the avoidance of doubt, any narrower definition of a substituent offered above can be combined with any other broad or narrowed definition of any other substituent. Throughout the disclosure above, where a narrower definition of a substituent is presented, that narrower definition is deemed disclosed in conjunction with all broader and narrower definitions of other substituents in the application. Intermediates Whilst the invention primarily relates to catalysts complexes, it will be appreciated that the ligands used to form those complexes are also new. The novel indenes of the present invention bear the combination of the distinctive features of the metallocene ligand framework: 1: 5-alkoxy indene, preferably methoxy indene, with 6-tertiary hydrocarbyl, preferably tertiary alkyl, substituent, and 2: an alpha-branched alkyl substituent on the 2-position of the indene.
The present invention accordingly further relates to indenes of formula (II)
 With: w25 (polymer weight) and V25 (Volume of TCB at 25°C). The column set was calibrated using universal calibration (according to ISO 16014-2:2019) with 19 narrow MWD polystyrene (PS) standards in the range of 0.5 kg/mol to 11500 kg/mol. The PS standards were dissolved at 160°C for 15 min or alternatively at room temperatures at a concentration of 0.2 mg/ml for molecular weight higher and equal 899 kg/mol and at a concentration of 1 mg/ml for molecular weight below 899 kg/mol. The conversion of the polystyrene peak molecular weight to polypropylene molecular weights was accomplished by using the Mark Houwink equation and the following Mark Houwink constants: KPS = 19 x 10-5 ml/g, αPS = 0.655 KPP = 39 x 10-5 ml/g, αPP = 0.725 A third order polynomial fit was used to fit the calibration data. All samples were prepared in the concentration range of 0.5 -1 mg/ml and dissolved at 160 °C for 3 hours under continuous gentle shaking Molecular weight averages (Mn, Mw, Mv and Mz), Molecular weight distribution (MWD) and its broadness, described by the polydispersity index PD= Mw/Mn (wherein Mn is the number average molecular weight and Mw is the weight average molecular weight) were determined using the following formulas:
With: w25 (polymer weight) and V25 (Volume of TCB at 25°C). The column set was calibrated using universal calibration (according to ISO 16014-2:2019) with 19 narrow MWD polystyrene (PS) standards in the range of 0.5 kg/mol to 11500 kg/mol. The PS standards were dissolved at 160°C for 15 min or alternatively at room temperatures at a concentration of 0.2 mg/ml for molecular weight higher and equal 899 kg/mol and at a concentration of 1 mg/ml for molecular weight below 899 kg/mol. The conversion of the polystyrene peak molecular weight to polypropylene molecular weights was accomplished by using the Mark Houwink equation and the following Mark Houwink constants: KPS = 19 x 10-5 ml/g, αPS = 0.655 KPP = 39 x 10-5 ml/g, αPP = 0.725 A third order polynomial fit was used to fit the calibration data. All samples were prepared in the concentration range of 0.5 -1 mg/ml and dissolved at 160 °C for 3 hours under continuous gentle shaking Molecular weight averages (Mn, Mw, Mv and Mz), Molecular weight distribution (MWD) and its broadness, described by the polydispersity index PD= Mw/Mn (wherein Mn is the number average molecular weight and Mw is the weight average molecular weight) were determined using the following formulas:

 The DSC curves and data have been produced on a DSC Q200 TA Instrument, by placing a 5-7 mg sample cut from the polymer MFR string, into a closed DSC aluminum pan, heating the sample from -10 °C to 225 °C at 10 °C/min, holding for 10 min at 225 °C, cooling from 225 °C to –30 °C, holding for 5 min at –30 °C, heating from –30 °C to 225 °C at 10 °C/min. The reported Tm values are those of the peak of the endothermic heat flow determined from the second heating scan. Melt Flow Rate The melt flow rate (MFR) was determined according to ISO 1133 and is indicated in g/10 min. The MFR is an indication of the flowability, and hence the processability, of the polymer. The higher the melt flow rate, the lower the molecular weight of the polymer. The MFR was determined at 230°C and may be determined at different loadings such as 2.16 kg (MFR2) or 21.6 kg (MFR21). Comonomer content by FTIR Quantitative infrared (IR) spectroscopy was used to estimate the C2 content of the copolymers through calibration to a primary method (NMR spectroscopy). Calibration was facilitated through the use of a set of in-house, non-commercial calibration standards of known C2 contents determined by quantitative 13C solution-state nuclear magnetic resonance (NMR) spectroscopy. The calibration procedure was undertaken in the conventional manner well documented in the literature [Spectroscopy of Polymers, 2nd Edition, J.L. Koenig, Elsevier Science, 1999]. The calibration set consisted of 8 calibration standards with C2 contents ranging between 0.0-3.5 wt%. Quantitative FTIR spectra were recorded in the solid-state using a Bruker Vertex 70 FTIR spectrometer. Spectra were recorded on 25x25 mm square films of 300 µm thickness prepared by compression moulding at 180 - 210°C and 70 bar of pressure. Standard transmission FTIR spectroscopy was employed using a spectral range of 5000-400 cm-1, an aperture of 6 mm, a spectral resolution of 2 cm-1, 16 background scans, 16 spectrum scans, an interferogram zero filling factor of 32 and Norton Beer strong apodisation.
Quantitative analysis was carried out by integration of (peak height) CH2 rocking deformations at 732.5 cm-1 (AQ) corresponding to isolated ethylene incorporation in PEP comonomer sequences (integration method K-OPUS, limits 759 and 702 cm-1). The quantitative band was normalized to the height of the CH combination band at 4323 cm-1 (AR) corresponding to CH structural units (integration method K, limits 4480, 3950 cm-1). The C2 content in units of weight percent was then predicted from the normalized absorption (A0 = AQ / AR) using linear calibration curve. The calibration curve having previously been constructed by least square regression of the normalized absorptions and comonomer contents measured by the primary technique (NMR Spectroscopy). The typical linear calibration curve has the form: ^ = ^^ × ^^ + ^^ Equation 1 Here C1 is the slope of the calibration curve with 0.96 > C1 > 1, and the intercept of -0.02 > C0 > 0.06. The quality of the calibration was assessed using the usual Confidence of Determination (COD) or R2. The COD was around 0.998. NMR Quantitative nuclear-magnetic resonance (NMR) spectroscopy was used to quantify the isotacticity and content of regio-defects of the polypropylene homopolymers. Quantitative 13C{1H} NMR spectra recorded in the solution-state using a Bruker Avance III 400 NMR spectrometer operating at 400.15 and 100.62 MHz for 1H and 13C respectively. All spectra were recorded using a 13C optimised 10 mm selective excitation probehead at 125°C using nitrogen gas for all pneumatics. Approximately 200 mg of material was dissolved in 1,2- tetrachloroethane-d2 (TCE-d2). This setup was chosen primarily for the high resolution needed for tacticity distribution quantification (Busico, V., Cipullo, R., Prog. Polym. Sci.26 (2001) 443; Busico, V.; Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). Standard single-pulse excitation was employed utilising the NOE and bi-level WALTZ16 decoupling scheme (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, B., J. Mag. Reson.187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28, 11289). A total of 6144 (6k) transients were acquired per spectra using a 3 s recycle delay. Quantitative 13C{1H} NMR spectra were processed, integrated and relevant quantitative properties determined from the integrals using proprietary computer programs. All chemical shifts are internally referenced to the methyl signal of the isotactic pentad mmmm at 21.85 ppm.
The tacticity distribution was quantified through integration of the methyl region between 23.6 and 19.7 ppm correcting for any sites not related to the stereo sequences of interest (Busico, V., Cipullo, R., Prog. Polym. Sci.26 (2001) 443; Busico, V., Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). The pentad isotacticity was determined through direct integration of the methyl region and reported as either the mole fraction or percentage of isotactic pentad mmmm with respect to all steric pentads i.e. [mmmm] = mmmm / sum of all steric pentads. When appropriate integrals were corrected for the presence of sites not directly associated with steric pentads. Characteristic signals corresponding to regio irregular propene insertion were observed (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253). The presence of secondary inserted propene in the form of 2,1 erythro regio defects was indicated by the presence of the two methyl signals at 17.7 and 17.2 ppm and confirmed by the presence of other characteristic signals. The amount of 2,1 erythro regio defects was quantified using the average integral (e) of the e6 and e8 sites observed at 17.7 and 17.2 ppm respectively, i.e. e = 0.5 * (e6 + e8). Characteristic signals corresponding to other types of regio irregularity were not observed (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253). The amount of primary inserted propene (p) was quantified based on the integral of all signals in the methyl region (CH3) from 23.6 to 19.7 ppm paying attention to correct for other species included in the integral not related to primary insertion and for primary insertion signals excluded from this region such that p = CH3 + 2*e. The relative content of a specific type of regio defect was reported as the mole fraction or percentage of said regio defect with respect all observed forms of propene insertion i.e. sum of all primary (1,2), secondary (2,1) and tertiary (3,1) inserted propene units, e.g. [21e] = e / ( p + e + t + i ). The total amount of secondary inserted propene in the form of 2,1-erythro or 2,1-threo regio defects was quantified as sum of all said regio irregular units, i.e. [21] = [21e] + [21t]. Quantitative nuclear-magnetic resonance (NMR) spectroscopy was used to quantify the ethylene content and the isotacticity of the copolymers. Quantitative 13C{1H} NMR spectra were recorded in the solution-state using a Bruker Avance III 400 NMR spectrometer operating at 400.15 and 100.62 MHz for 1H and 13C respectively. All spectra were recorded using a 13C optimised 10 mm extended temperature probehead at 125°C using nitrogen gas for all pneumatics. Approximately 200 mg of material was dissolved in 3 ml of 1,2-tetrachloroethane-d2 (TCE-d2) along with chromium-(III)-acetylacetonate (Cr(acac)3) resulting in a 65 mM solution of relaxation
agent in solvent as described in G. Singh, A. Kothari, V. Gupta, Polymer Testing 2009, 28(5), 475. To ensure a homogenous solution, after initial sample preparation in a heat block, the NMR tube was further heated in a rotatory oven for at least 1 hour. Upon insertion into the magnet the tube was spun at 10 Hz. This setup was chosen primarily for the high resolution and quantitatively needed for accurate ethylene content quantification. Standard single- pulse excitation was employed without NOE, using an optimised tip angle, 1 s recycle delay and a bi-level WALTZ16 decoupling scheme as described in Z. Zhou, R. Kuemmerle, X. Qiu, D. Redwine, R. Cong, A. Taha, D. Baugh, B. Winniford, J. Mag. Reson.187 (2007) 225 and V. Busico, P. Carbonniere, R. Cipullo, C. Pellecchia, J. Severn, G. Talarico, Macromol. Rapid Commun.2007, 28, 1128. A total of 6144 (6k) transients were acquired per spectra. Quantitative 13C{1H} NMR spectra were processed, integrated and relevant quantitative properties determined from the integrals. All chemical shifts were indirectly referenced to the central methylene group of the ethylene block (EEE) at 30.00 ppm using the chemical shift of the solvent. This approach allowed comparable referencing even when this structural unit was not present. With characteristic signals corresponding to 2,1 erythro regiodefects observed (as described in L. Resconi, L. Cavallo, A. Fait, F. Piemontesi, Chem. Rev. 2000, 100 (4), 1253, in Cheng, H. N., Macromolecules 1984, 17, 1950, and in W-J. Wang and S. Zhu, Macromolecules 2000, 33, 1157) the correction for the influence of the regiodefects on determined properties was required. Characteristic signals corresponding to other types of regiodefects were not observed. Characteristic signals corresponding to the incorporation of ethylene were observed (as described in Cheng, H. N., Macromolecules 1984, 17, 1950) and the comonomer fraction calculated as the fraction of ethylene in the polymer with respect to all monomer in the polymer: fE = ( E / ( P + E ) The comonomer fraction was quantified using the method of W-J. Wang and S. Zhu, Macromolecules 2000, 33, 1157, through integration of multiple signals across the whole spectral region in the 13C{1H} spectra. This method was chosen for its robust nature and ability to account for the presence of regio-defects when needed. Integral regions were slightly adjusted to increase applicability across the whole range of encountered comonomer contents. The mole percent comonomer incorporation was calculated from the mole fraction:
E [mol%] = 100 * fE The weight percent comonomer incorporation was calculated from the mole fraction: E [wt%] = 100 * ( fE * 28.06 ) / ( (fE * 28.06) + ((1-fE) * 42.08) ) The isotacticity of the copolymer was determined according to known methods, for example as described in Macromolecules 2005, vol.38, pp.3054-3059. Crystex The crystalline (CF) and soluble fractions (SF) of the heterophasic propylene resins as well as the comonomer content and intrinsic viscosities of the respective fractions were analysed by the Crystex method. The crystalline and amorphous fractions are separated through temperature cycles of dissolution at 160°C, crystallization at 40°C and re- dissolution in 1,2,4-trichlorobenzene (1,2,4-TCB) at 160°C. Quantification of SF and CF and determination of ethylene content (C2) were achieved by means of an infrared detector (IR4) and an online 2-capillary viscometer was used for determination of the intrinsic viscosity (iV). IR4 detector is multiple wavelength detector detecting IR absorbance at two different bands (CH3 and CH2) for the determination of the concentration and the ethylene content in ethylene-propylene copolymers. IR4 detector is calibrated with series of EP copolymers with known ethylene content in the range of 2 wt.-% to 69 wt.-% (determined by 13C- NMR). Amount of Soluble fraction (SF) and Crystalline Fraction (CF) are correlated through the XS calibration to the “Xylene Soluble” (XS) quantity and respectively Xylene Insoluble (XI) fractions, determined according to standard gravimetric method as per ISO16152 (2005). XS calibration is achieved by testing various EP copolymers with XS content in the range 2-31 wt%. Intrinsic viscosity (iV) of the parent EP copolymer and its soluble and crystalline fractions are determined with a use of an online 2-capillary viscometer and are correlated to corresponding iV determined in decalin according to ISO 1628-3 (2010). Calibration is achieved with several commercial EP PP copolymers with iV = 2-4 dL/g. A sample of the PP composition to be analysed is weighed out in concentrations of 10mg/ml to 20mg/ml. After automated filling of the vial with 1,2,4-TCB containing 250 mg/l 2,6-tert-butyl-4-methylphenol (BHT) as antioxidant, the sample is dissolved at 160°C until complete dissolution is achieved, usually for 60 min, with constant stirring of 800rpm. A defined volume of the sample solution is injected into the column filled with inert support where the crystallization of the sample and separation of the soluble fraction from the
crystalline part is taking place. This process is repeated two times. During the first injection the whole sample is measured at elevated temperature, determining the iV[dl/g] and the C2[wt%] of the PP composition. During the second injection the soluble fraction (at low temperature) and the crystalline fraction (at elevated temperature) with the crystallization cycle are determined (wt% SF, wt% C2, iV). Metallocene synthesis Synthesis of MC-CE1 MC-CE1 was prepared as described in WO2001048034, metallocene example 18. Synthesis of MC-CE2 MC-CE2, was prepared as described in WO2005058916, metallocene example 1. Synthesis of MC-CE3 Rac-dimethylsilanediyl-bis[2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert- butyl-inden- l-yl]zirconium dichloride, MC-CE3, was prepared as described in WO2018091684 for MC- IE1. Synthesis of MC-CE4 Rac-dimethylsilanediyl-bis[2-neopentyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert- butylinden-1–yl]zirconium dichloride, was prepared as described in WO2018091684 for MC-IE4. Synthesis of MC-IE1 and MC-IE2 Isopropylmalonic acid
The DSC curves and data have been produced on a DSC Q200 TA Instrument, by placing a 5-7 mg sample cut from the polymer MFR string, into a closed DSC aluminum pan, heating the sample from -10 °C to 225 °C at 10 °C/min, holding for 10 min at 225 °C, cooling from 225 °C to –30 °C, holding for 5 min at –30 °C, heating from –30 °C to 225 °C at 10 °C/min. The reported Tm values are those of the peak of the endothermic heat flow determined from the second heating scan. Melt Flow Rate The melt flow rate (MFR) was determined according to ISO 1133 and is indicated in g/10 min. The MFR is an indication of the flowability, and hence the processability, of the polymer. The higher the melt flow rate, the lower the molecular weight of the polymer. The MFR was determined at 230°C and may be determined at different loadings such as 2.16 kg (MFR2) or 21.6 kg (MFR21). Comonomer content by FTIR Quantitative infrared (IR) spectroscopy was used to estimate the C2 content of the copolymers through calibration to a primary method (NMR spectroscopy). Calibration was facilitated through the use of a set of in-house, non-commercial calibration standards of known C2 contents determined by quantitative 13C solution-state nuclear magnetic resonance (NMR) spectroscopy. The calibration procedure was undertaken in the conventional manner well documented in the literature [Spectroscopy of Polymers, 2nd Edition, J.L. Koenig, Elsevier Science, 1999]. The calibration set consisted of 8 calibration standards with C2 contents ranging between 0.0-3.5 wt%. Quantitative FTIR spectra were recorded in the solid-state using a Bruker Vertex 70 FTIR spectrometer. Spectra were recorded on 25x25 mm square films of 300 µm thickness prepared by compression moulding at 180 - 210°C and 70 bar of pressure. Standard transmission FTIR spectroscopy was employed using a spectral range of 5000-400 cm-1, an aperture of 6 mm, a spectral resolution of 2 cm-1, 16 background scans, 16 spectrum scans, an interferogram zero filling factor of 32 and Norton Beer strong apodisation.
Quantitative analysis was carried out by integration of (peak height) CH2 rocking deformations at 732.5 cm-1 (AQ) corresponding to isolated ethylene incorporation in PEP comonomer sequences (integration method K-OPUS, limits 759 and 702 cm-1). The quantitative band was normalized to the height of the CH combination band at 4323 cm-1 (AR) corresponding to CH structural units (integration method K, limits 4480, 3950 cm-1). The C2 content in units of weight percent was then predicted from the normalized absorption (A0 = AQ / AR) using linear calibration curve. The calibration curve having previously been constructed by least square regression of the normalized absorptions and comonomer contents measured by the primary technique (NMR Spectroscopy). The typical linear calibration curve has the form: ^ = ^^ × ^^ + ^^ Equation 1 Here C1 is the slope of the calibration curve with 0.96 > C1 > 1, and the intercept of -0.02 > C0 > 0.06. The quality of the calibration was assessed using the usual Confidence of Determination (COD) or R2. The COD was around 0.998. NMR Quantitative nuclear-magnetic resonance (NMR) spectroscopy was used to quantify the isotacticity and content of regio-defects of the polypropylene homopolymers. Quantitative 13C{1H} NMR spectra recorded in the solution-state using a Bruker Avance III 400 NMR spectrometer operating at 400.15 and 100.62 MHz for 1H and 13C respectively. All spectra were recorded using a 13C optimised 10 mm selective excitation probehead at 125°C using nitrogen gas for all pneumatics. Approximately 200 mg of material was dissolved in 1,2- tetrachloroethane-d2 (TCE-d2). This setup was chosen primarily for the high resolution needed for tacticity distribution quantification (Busico, V., Cipullo, R., Prog. Polym. Sci.26 (2001) 443; Busico, V.; Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). Standard single-pulse excitation was employed utilising the NOE and bi-level WALTZ16 decoupling scheme (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, B., J. Mag. Reson.187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28, 11289). A total of 6144 (6k) transients were acquired per spectra using a 3 s recycle delay. Quantitative 13C{1H} NMR spectra were processed, integrated and relevant quantitative properties determined from the integrals using proprietary computer programs. All chemical shifts are internally referenced to the methyl signal of the isotactic pentad mmmm at 21.85 ppm.
The tacticity distribution was quantified through integration of the methyl region between 23.6 and 19.7 ppm correcting for any sites not related to the stereo sequences of interest (Busico, V., Cipullo, R., Prog. Polym. Sci.26 (2001) 443; Busico, V., Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). The pentad isotacticity was determined through direct integration of the methyl region and reported as either the mole fraction or percentage of isotactic pentad mmmm with respect to all steric pentads i.e. [mmmm] = mmmm / sum of all steric pentads. When appropriate integrals were corrected for the presence of sites not directly associated with steric pentads. Characteristic signals corresponding to regio irregular propene insertion were observed (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253). The presence of secondary inserted propene in the form of 2,1 erythro regio defects was indicated by the presence of the two methyl signals at 17.7 and 17.2 ppm and confirmed by the presence of other characteristic signals. The amount of 2,1 erythro regio defects was quantified using the average integral (e) of the e6 and e8 sites observed at 17.7 and 17.2 ppm respectively, i.e. e = 0.5 * (e6 + e8). Characteristic signals corresponding to other types of regio irregularity were not observed (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253). The amount of primary inserted propene (p) was quantified based on the integral of all signals in the methyl region (CH3) from 23.6 to 19.7 ppm paying attention to correct for other species included in the integral not related to primary insertion and for primary insertion signals excluded from this region such that p = CH3 + 2*e. The relative content of a specific type of regio defect was reported as the mole fraction or percentage of said regio defect with respect all observed forms of propene insertion i.e. sum of all primary (1,2), secondary (2,1) and tertiary (3,1) inserted propene units, e.g. [21e] = e / ( p + e + t + i ). The total amount of secondary inserted propene in the form of 2,1-erythro or 2,1-threo regio defects was quantified as sum of all said regio irregular units, i.e. [21] = [21e] + [21t]. Quantitative nuclear-magnetic resonance (NMR) spectroscopy was used to quantify the ethylene content and the isotacticity of the copolymers. Quantitative 13C{1H} NMR spectra were recorded in the solution-state using a Bruker Avance III 400 NMR spectrometer operating at 400.15 and 100.62 MHz for 1H and 13C respectively. All spectra were recorded using a 13C optimised 10 mm extended temperature probehead at 125°C using nitrogen gas for all pneumatics. Approximately 200 mg of material was dissolved in 3 ml of 1,2-tetrachloroethane-d2 (TCE-d2) along with chromium-(III)-acetylacetonate (Cr(acac)3) resulting in a 65 mM solution of relaxation
agent in solvent as described in G. Singh, A. Kothari, V. Gupta, Polymer Testing 2009, 28(5), 475. To ensure a homogenous solution, after initial sample preparation in a heat block, the NMR tube was further heated in a rotatory oven for at least 1 hour. Upon insertion into the magnet the tube was spun at 10 Hz. This setup was chosen primarily for the high resolution and quantitatively needed for accurate ethylene content quantification. Standard single- pulse excitation was employed without NOE, using an optimised tip angle, 1 s recycle delay and a bi-level WALTZ16 decoupling scheme as described in Z. Zhou, R. Kuemmerle, X. Qiu, D. Redwine, R. Cong, A. Taha, D. Baugh, B. Winniford, J. Mag. Reson.187 (2007) 225 and V. Busico, P. Carbonniere, R. Cipullo, C. Pellecchia, J. Severn, G. Talarico, Macromol. Rapid Commun.2007, 28, 1128. A total of 6144 (6k) transients were acquired per spectra. Quantitative 13C{1H} NMR spectra were processed, integrated and relevant quantitative properties determined from the integrals. All chemical shifts were indirectly referenced to the central methylene group of the ethylene block (EEE) at 30.00 ppm using the chemical shift of the solvent. This approach allowed comparable referencing even when this structural unit was not present. With characteristic signals corresponding to 2,1 erythro regiodefects observed (as described in L. Resconi, L. Cavallo, A. Fait, F. Piemontesi, Chem. Rev. 2000, 100 (4), 1253, in Cheng, H. N., Macromolecules 1984, 17, 1950, and in W-J. Wang and S. Zhu, Macromolecules 2000, 33, 1157) the correction for the influence of the regiodefects on determined properties was required. Characteristic signals corresponding to other types of regiodefects were not observed. Characteristic signals corresponding to the incorporation of ethylene were observed (as described in Cheng, H. N., Macromolecules 1984, 17, 1950) and the comonomer fraction calculated as the fraction of ethylene in the polymer with respect to all monomer in the polymer: fE = ( E / ( P + E ) The comonomer fraction was quantified using the method of W-J. Wang and S. Zhu, Macromolecules 2000, 33, 1157, through integration of multiple signals across the whole spectral region in the 13C{1H} spectra. This method was chosen for its robust nature and ability to account for the presence of regio-defects when needed. Integral regions were slightly adjusted to increase applicability across the whole range of encountered comonomer contents. The mole percent comonomer incorporation was calculated from the mole fraction:
E [mol%] = 100 * fE The weight percent comonomer incorporation was calculated from the mole fraction: E [wt%] = 100 * ( fE * 28.06 ) / ( (fE * 28.06) + ((1-fE) * 42.08) ) The isotacticity of the copolymer was determined according to known methods, for example as described in Macromolecules 2005, vol.38, pp.3054-3059. Crystex The crystalline (CF) and soluble fractions (SF) of the heterophasic propylene resins as well as the comonomer content and intrinsic viscosities of the respective fractions were analysed by the Crystex method. The crystalline and amorphous fractions are separated through temperature cycles of dissolution at 160°C, crystallization at 40°C and re- dissolution in 1,2,4-trichlorobenzene (1,2,4-TCB) at 160°C. Quantification of SF and CF and determination of ethylene content (C2) were achieved by means of an infrared detector (IR4) and an online 2-capillary viscometer was used for determination of the intrinsic viscosity (iV). IR4 detector is multiple wavelength detector detecting IR absorbance at two different bands (CH3 and CH2) for the determination of the concentration and the ethylene content in ethylene-propylene copolymers. IR4 detector is calibrated with series of EP copolymers with known ethylene content in the range of 2 wt.-% to 69 wt.-% (determined by 13C- NMR). Amount of Soluble fraction (SF) and Crystalline Fraction (CF) are correlated through the XS calibration to the “Xylene Soluble” (XS) quantity and respectively Xylene Insoluble (XI) fractions, determined according to standard gravimetric method as per ISO16152 (2005). XS calibration is achieved by testing various EP copolymers with XS content in the range 2-31 wt%. Intrinsic viscosity (iV) of the parent EP copolymer and its soluble and crystalline fractions are determined with a use of an online 2-capillary viscometer and are correlated to corresponding iV determined in decalin according to ISO 1628-3 (2010). Calibration is achieved with several commercial EP PP copolymers with iV = 2-4 dL/g. A sample of the PP composition to be analysed is weighed out in concentrations of 10mg/ml to 20mg/ml. After automated filling of the vial with 1,2,4-TCB containing 250 mg/l 2,6-tert-butyl-4-methylphenol (BHT) as antioxidant, the sample is dissolved at 160°C until complete dissolution is achieved, usually for 60 min, with constant stirring of 800rpm. A defined volume of the sample solution is injected into the column filled with inert support where the crystallization of the sample and separation of the soluble fraction from the
crystalline part is taking place. This process is repeated two times. During the first injection the whole sample is measured at elevated temperature, determining the iV[dl/g] and the C2[wt%] of the PP composition. During the second injection the soluble fraction (at low temperature) and the crystalline fraction (at elevated temperature) with the crystallization cycle are determined (wt% SF, wt% C2, iV). Metallocene synthesis Synthesis of MC-CE1 MC-CE1 was prepared as described in WO2001048034, metallocene example 18. Synthesis of MC-CE2 MC-CE2, was prepared as described in WO2005058916, metallocene example 1. Synthesis of MC-CE3 Rac-dimethylsilanediyl-bis[2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert- butyl-inden- l-yl]zirconium dichloride, MC-CE3, was prepared as described in WO2018091684 for MC- IE1. Synthesis of MC-CE4 Rac-dimethylsilanediyl-bis[2-neopentyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert- butylinden-1–yl]zirconium dichloride, was prepared as described in WO2018091684 for MC-IE4. Synthesis of MC-IE1 and MC-IE2 Isopropylmalonic acid
 Diethylamine (62.4 ml, 4.3 g, 0.606 mol) was added dropwise at 5 °C to a solution of isopropylmalonic acid (76.4 g, 523 mmol) in 750 ml of ethyl acetate. Paraform (22.1 g, 0.736 mol) was added to the obtained suspension. The resulting mixture was refluxed for 5 h, then cooled to 5 °C, and 350 ml of ether and 1000 cm3 of 2 M HCl were added. After mixing, the organic layer was separated, the aqueous layer was additionally extracted with 2 ^500 ml of ether. The combined organic phase was dried over Na2SO4 and then evaporated to dryness. The residue was purified by vacuum distillation to give 2- isopropylacrylic acid, bp 65 °C/4 mm Hg. Yield 57.0 g (95.5%) of a colorless liquid. 1H NMR (CDCl3): δ 12.45 (br.s, 1H), 6.30 (s, 1H), 5.65 (t, 1H), 2.81 (septd, J = 6.9 Hz, J = 0.9 Hz, 1H), 1.11 (d, J = 6.9 Hz, 6H).13C NMR (CDCl3): δ 173.30, 146.47, 124.31, 28.94, 21.76. 6-tert-Butyl-5-methoxy-2-isopropylindan-1-one
Diethylamine (62.4 ml, 4.3 g, 0.606 mol) was added dropwise at 5 °C to a solution of isopropylmalonic acid (76.4 g, 523 mmol) in 750 ml of ethyl acetate. Paraform (22.1 g, 0.736 mol) was added to the obtained suspension. The resulting mixture was refluxed for 5 h, then cooled to 5 °C, and 350 ml of ether and 1000 cm3 of 2 M HCl were added. After mixing, the organic layer was separated, the aqueous layer was additionally extracted with 2 ^500 ml of ether. The combined organic phase was dried over Na2SO4 and then evaporated to dryness. The residue was purified by vacuum distillation to give 2- isopropylacrylic acid, bp 65 °C/4 mm Hg. Yield 57.0 g (95.5%) of a colorless liquid. 1H NMR (CDCl3): δ 12.45 (br.s, 1H), 6.30 (s, 1H), 5.65 (t, 1H), 2.81 (septd, J = 6.9 Hz, J = 0.9 Hz, 1H), 1.11 (d, J = 6.9 Hz, 6H).13C NMR (CDCl3): δ 173.30, 146.47, 124.31, 28.94, 21.76. 6-tert-Butyl-5-methoxy-2-isopropylindan-1-one
 114.1 g (1.0 mol) of 2-isopropylacrylic acid was added to Eaton's reagent obtained from 220 g of P4O10 and 1120 ml of MeSO3H at 50 °C. 131.2 g (0.8 mol) of 1-tert-butyl-2- methoxybenzene was added dropwise to this mixture with vigorous stirring for ca.1 h at 50-53 °C (water bath temperature). The resulting mixture was stirred for 1 h at this temperature, then cooled to room temperature, and poured on a mixture of 1.5 liter of cold water and 3 kg of ice. The crude product was extracted with 3 ^600 ml of dichloromethane. The combined organic phase was washed by aqueous K2CO3, dried over K2CO3, filtered through a short pad of silica gel 60 (40-63 µm) and then evaporated to dryness. The residue was purified by vacuum distillation to give 193.8 g (93.0 %, ca.95% purity) of 6- tert-butyl-5-methoxy-2-isopropylindan-1-one as a yellowish oil (bp 150-190oC /4 mm Hg).
1H NMR (CDCl3): δ 7.66 (s, 1H), 6.89 (s, 1H), 3.93 (s, 3H), 3.04 (dd, J = 17.4 Hz, J = 8.0 Hz, 1H), 2.84 (dd, J = 17.4 Hz, J = 3.8 Hz, 1H), 2.67-2.60 (m, 1H), 2.48-2.34 (m, 1H), 1.37 (s, 9H), 1.05 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.9 Hz, 3H). 13C NMR (CDCl3): δ 207.45, 164.44, 155.10, 138.54, 130.07, 121.69, 107.64, 55.14, 53.20, 35.00, 29.54, 28.87, 27.65, 20.96, 17.00. 4-Bromo-6-tert-butyl-2-isopropyl-5-methoxyindan-1-one
114.1 g (1.0 mol) of 2-isopropylacrylic acid was added to Eaton's reagent obtained from 220 g of P4O10 and 1120 ml of MeSO3H at 50 °C. 131.2 g (0.8 mol) of 1-tert-butyl-2- methoxybenzene was added dropwise to this mixture with vigorous stirring for ca.1 h at 50-53 °C (water bath temperature). The resulting mixture was stirred for 1 h at this temperature, then cooled to room temperature, and poured on a mixture of 1.5 liter of cold water and 3 kg of ice. The crude product was extracted with 3 ^600 ml of dichloromethane. The combined organic phase was washed by aqueous K2CO3, dried over K2CO3, filtered through a short pad of silica gel 60 (40-63 µm) and then evaporated to dryness. The residue was purified by vacuum distillation to give 193.8 g (93.0 %, ca.95% purity) of 6- tert-butyl-5-methoxy-2-isopropylindan-1-one as a yellowish oil (bp 150-190oC /4 mm Hg).
1H NMR (CDCl3): δ 7.66 (s, 1H), 6.89 (s, 1H), 3.93 (s, 3H), 3.04 (dd, J = 17.4 Hz, J = 8.0 Hz, 1H), 2.84 (dd, J = 17.4 Hz, J = 3.8 Hz, 1H), 2.67-2.60 (m, 1H), 2.48-2.34 (m, 1H), 1.37 (s, 9H), 1.05 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.9 Hz, 3H). 13C NMR (CDCl3): δ 207.45, 164.44, 155.10, 138.54, 130.07, 121.69, 107.64, 55.14, 53.20, 35.00, 29.54, 28.87, 27.65, 20.96, 17.00. 4-Bromo-6-tert-butyl-2-isopropyl-5-methoxyindan-1-one
 Bromine (20.8 ml, 64.9 g, 405.9 mmol) was added dropwise over 5 min to a mixture of 97.0 g (0.372 mol) of 6-tert-butyl-2-isopropyl-5-methoxyindan-1-one, 113.2 g of sodium acetate, 3.0 g of nBu4NI, 310 ml of dichloromethane, and 645 ml of water with vigorous stirring at 5 °C. This mixture was stirred at 5 °C for 2 h, then a solution of 52.2 g of sodium acetate in 290 ml of water was added followed by addition of 10.8 ml (33.7 g, 210.8 mmol) of bromine. The resulting mixture was additionally stirred for 1 h at this temperature and then washed by aqueous Na2SO3 to remove excess bromine. The crude product was extracted by 3 ^250 ml of dichloromethane. The combined organic extract was dried over K2CO3, evaporated to dryness, and the residue was dried in vacuum. This procedure gave 124.84 g (98.9%, ca. 95% purity) of a yellowish oil which was used without further purification. 1H NMR (CDCl3): δ 7.68 (s, 1H), 4.04 (s, 3H), 3.04 (dd, J = 17.9 Hz, J = 8.1 Hz, 1H), 2.80 (dd, J = 17.9 Hz, J = 3.9 Hz, 1H), 2.71-2.64 (m, 1H), 2.49-2.35 (m, 1H), 1.40 (s, 9H), 1.08 (d, J = 6.9 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H).13C NMR (CDCl3): δ 207.21, 162.59, 154.47, 145.24, 133.82, 121.03, 116.56, 61.54, 53.32, 35.56, 30.54, 29.41, 28.94, 20.78, 17.17. 6-tert-Butyl-2-isopropyl-5-methoxy-4-(3,5-dimethylphenyl)-indan-1-one
Bromine (20.8 ml, 64.9 g, 405.9 mmol) was added dropwise over 5 min to a mixture of 97.0 g (0.372 mol) of 6-tert-butyl-2-isopropyl-5-methoxyindan-1-one, 113.2 g of sodium acetate, 3.0 g of nBu4NI, 310 ml of dichloromethane, and 645 ml of water with vigorous stirring at 5 °C. This mixture was stirred at 5 °C for 2 h, then a solution of 52.2 g of sodium acetate in 290 ml of water was added followed by addition of 10.8 ml (33.7 g, 210.8 mmol) of bromine. The resulting mixture was additionally stirred for 1 h at this temperature and then washed by aqueous Na2SO3 to remove excess bromine. The crude product was extracted by 3 ^250 ml of dichloromethane. The combined organic extract was dried over K2CO3, evaporated to dryness, and the residue was dried in vacuum. This procedure gave 124.84 g (98.9%, ca. 95% purity) of a yellowish oil which was used without further purification. 1H NMR (CDCl3): δ 7.68 (s, 1H), 4.04 (s, 3H), 3.04 (dd, J = 17.9 Hz, J = 8.1 Hz, 1H), 2.80 (dd, J = 17.9 Hz, J = 3.9 Hz, 1H), 2.71-2.64 (m, 1H), 2.49-2.35 (m, 1H), 1.40 (s, 9H), 1.08 (d, J = 6.9 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H).13C NMR (CDCl3): δ 207.21, 162.59, 154.47, 145.24, 133.82, 121.03, 116.56, 61.54, 53.32, 35.56, 30.54, 29.41, 28.94, 20.78, 17.17. 6-tert-Butyl-2-isopropyl-5-methoxy-4-(3,5-dimethylphenyl)-indan-1-one
 A mixture of 124.84 g (368.0 mmol) of 4-bromo-6-tert-butyl-2-isopropyl-5-methoxyindan- 1-one, 69.7 g (464.7 mmol, 1.26 equiv.) of 3,5-Me2C6H3B(OH)2, 1.9 g (3.72 mmol, 1 mol. %) of Pd(PtBu3)2, 118.3 g of Na2CO3, 600 ml of 2-methyltetrahydrofurane and 540 ml of water was refluxed for 6 h. Then, 500 ml of water was added, the organic layer was separated, and the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic extract was dried over K2CO3 and then evaporated to dryness to give slightly yellowish solid mass. The product was isolated by flash-chromatography on silica gel 60 (40-63 µm, eluent: hexanes-dichloromethane = 1:1 and then 1:5, vol.). Yield 121.55 g (90.6%, purity ca.95%) of a yellowish crystalline material. 1H NMR (CDCl3): δ 7.71 (s, 1H), 7.04 (s, 1H), 7.03 (s, 2H), 3.31 (s, 3H), 2.87 (dd, J = 18.5 Hz, J = 8.8 Hz, 1H), 2.65-2.54 (dd and m, 2H), 2.43-2.34 (s and m, 7H), 1.42 (s, 9H), 0.99 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.8 Hz, 3H).13C NMR (CDCl3): δ 208.19, 163.34, 153.44, 143.11, 138.08, 136.33, 132.64, 132.12, 129.08, 127.20, 120.93, 77.00, 60.46, 53.37, 35.33, 30.50, 28.88, 27.38, 21.39, 20.90, 17.25. 5-tert-Butyl-2-isopropyl-6-methoxy-7-(3,5-dimethylphenyl)-1H-indene
A mixture of 124.84 g (368.0 mmol) of 4-bromo-6-tert-butyl-2-isopropyl-5-methoxyindan- 1-one, 69.7 g (464.7 mmol, 1.26 equiv.) of 3,5-Me2C6H3B(OH)2, 1.9 g (3.72 mmol, 1 mol. %) of Pd(PtBu3)2, 118.3 g of Na2CO3, 600 ml of 2-methyltetrahydrofurane and 540 ml of water was refluxed for 6 h. Then, 500 ml of water was added, the organic layer was separated, and the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic extract was dried over K2CO3 and then evaporated to dryness to give slightly yellowish solid mass. The product was isolated by flash-chromatography on silica gel 60 (40-63 µm, eluent: hexanes-dichloromethane = 1:1 and then 1:5, vol.). Yield 121.55 g (90.6%, purity ca.95%) of a yellowish crystalline material. 1H NMR (CDCl3): δ 7.71 (s, 1H), 7.04 (s, 1H), 7.03 (s, 2H), 3.31 (s, 3H), 2.87 (dd, J = 18.5 Hz, J = 8.8 Hz, 1H), 2.65-2.54 (dd and m, 2H), 2.43-2.34 (s and m, 7H), 1.42 (s, 9H), 0.99 (d, J = 6.9 Hz, 3H), 0.77 (d, J = 6.8 Hz, 3H).13C NMR (CDCl3): δ 208.19, 163.34, 153.44, 143.11, 138.08, 136.33, 132.64, 132.12, 129.08, 127.20, 120.93, 77.00, 60.46, 53.37, 35.33, 30.50, 28.88, 27.38, 21.39, 20.90, 17.25. 5-tert-Butyl-2-isopropyl-6-methoxy-7-(3,5-dimethylphenyl)-1H-indene
 NaBH4 (18.9 g, 0.5 mol, 1.5 equiv.) was added to a solution of 121.55 g (333.45 mmol) of 6-tert-butyl-2-isopropyl-5-methoxy-4-(3,5-dimethylphenyl)-indan-1-one in 600 ml of THF cooled to 5 °C. MeOH (300 ml) was added dropwise to this mixture over ca.5 h at 5 °C, and the resulting mixture was stirred overnight at room temperature. Then, this mixture was evaporated to dryness, 1000 ml of dichloromethane and 1000 ml water were added to the residue, and the so obtained mixture was acidified with 2 M HCl to pH~6.5. The organic layer was separated, the aqueous layer was additionally extracted with 100 ml of dichloromethane. The combined organic phase was passed through a pad (~30 ml) of silica gel 60 (40-63 µm; eluent: dichloromethane) to get rid of most of the palladium black. The obtained elute was evaporated to dryness to give a grey solid mass. To a solution of this mass in 1000 ml of toluene 1.0 g of TsOH was added. This mixture was refluxed with Dean-Stark head for 10 min and then cooled to room temperature using water bath. The formed solution was washed with 10% Na2CO3, the organic layer was separated, the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic
phase was dried over K2CO3 and then evaporated to dryness. The crude product was purified by flash chromatography on silica gel 60 (40-63 µm, hexanes-dichloromethane = 5:1) followed by recrystallization from n-hexane (hot→ –30 °C). This procedure gave 96.93 g (83.4%) of pure 5-tert-butyl-7-(3,5-dimethylphenyl)-2-isopropyl-6-methoxy-1H-indene. 1H NMR (CDCl3): δ 7.24 (s, 1H), 7.09 (s, 2H), 6.99 (s, 1H), 6.46 (m, 1H), 3.24 (s, 3H), 3.15 (m, 2H), 2.68 (sept, J = 6.8 Hz, 1H), 2.37 (s, 6H), 1.43 (s, 9H), 1.14 (d, J = 6.8 Hz, 6H). 13C NMR (CDCl3): δ 156.43, 154.33, 141.34, 140.95, 140.23, 138.29, 137.69, 131.99, 128.49, 127.22, 123.86, 117.32, 60.67, 39.15, 35.13, 31.00, 30.04, 22.64, 21.44. [6-tert-Butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-methyl-1H-inden-1-yl] chlorodimethylsilane
NaBH4 (18.9 g, 0.5 mol, 1.5 equiv.) was added to a solution of 121.55 g (333.45 mmol) of 6-tert-butyl-2-isopropyl-5-methoxy-4-(3,5-dimethylphenyl)-indan-1-one in 600 ml of THF cooled to 5 °C. MeOH (300 ml) was added dropwise to this mixture over ca.5 h at 5 °C, and the resulting mixture was stirred overnight at room temperature. Then, this mixture was evaporated to dryness, 1000 ml of dichloromethane and 1000 ml water were added to the residue, and the so obtained mixture was acidified with 2 M HCl to pH~6.5. The organic layer was separated, the aqueous layer was additionally extracted with 100 ml of dichloromethane. The combined organic phase was passed through a pad (~30 ml) of silica gel 60 (40-63 µm; eluent: dichloromethane) to get rid of most of the palladium black. The obtained elute was evaporated to dryness to give a grey solid mass. To a solution of this mass in 1000 ml of toluene 1.0 g of TsOH was added. This mixture was refluxed with Dean-Stark head for 10 min and then cooled to room temperature using water bath. The formed solution was washed with 10% Na2CO3, the organic layer was separated, the aqueous layer was extracted with 300 ml of dichloromethane. The combined organic
phase was dried over K2CO3 and then evaporated to dryness. The crude product was purified by flash chromatography on silica gel 60 (40-63 µm, hexanes-dichloromethane = 5:1) followed by recrystallization from n-hexane (hot→ –30 °C). This procedure gave 96.93 g (83.4%) of pure 5-tert-butyl-7-(3,5-dimethylphenyl)-2-isopropyl-6-methoxy-1H-indene. 1H NMR (CDCl3): δ 7.24 (s, 1H), 7.09 (s, 2H), 6.99 (s, 1H), 6.46 (m, 1H), 3.24 (s, 3H), 3.15 (m, 2H), 2.68 (sept, J = 6.8 Hz, 1H), 2.37 (s, 6H), 1.43 (s, 9H), 1.14 (d, J = 6.8 Hz, 6H). 13C NMR (CDCl3): δ 156.43, 154.33, 141.34, 140.95, 140.23, 138.29, 137.69, 131.99, 128.49, 127.22, 123.86, 117.32, 60.67, 39.15, 35.13, 31.00, 30.04, 22.64, 21.44. [6-tert-Butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-methyl-1H-inden-1-yl] chlorodimethylsilane
 nBuLi in hexanes (2.43 M, 14.6 ml, 35.5 mmol) was added in one portion to a solution of 5-tert-butyl-7-(3,5-dimethylphenyl)-6-methoxy-2-methyl-1H-indene (11.3 g, 35.3 mmol) in 200 ml of ether cooled to –50 °C. The resulting orange solution was stirred overnight at room temperature, then the obtained orange solution containing a yellowish precipitate was cooled to –78 °C (the precipitate almost completely disappeared), and dichlorodimethylsilane (22.8 g, 177 mmol, 5 equiv) was added in one portion. The formed solution was warmed to room temperature and stirred overnight at room temperature. The resulting mixture was filtered through glass frit (G4). The precipitate was additionally washed by 2×10 ml of ether. The combined filtrate was evaporated to dryness to give the title material as slightly orange oil which was used without further purification.
nBuLi in hexanes (2.43 M, 14.6 ml, 35.5 mmol) was added in one portion to a solution of 5-tert-butyl-7-(3,5-dimethylphenyl)-6-methoxy-2-methyl-1H-indene (11.3 g, 35.3 mmol) in 200 ml of ether cooled to –50 °C. The resulting orange solution was stirred overnight at room temperature, then the obtained orange solution containing a yellowish precipitate was cooled to –78 °C (the precipitate almost completely disappeared), and dichlorodimethylsilane (22.8 g, 177 mmol, 5 equiv) was added in one portion. The formed solution was warmed to room temperature and stirred overnight at room temperature. The resulting mixture was filtered through glass frit (G4). The precipitate was additionally washed by 2×10 ml of ether. The combined filtrate was evaporated to dryness to give the title material as slightly orange oil which was used without further purification.
 nBuLi in hexanes (2.5 M,13.1 ml, 32.75 mmol) was added in one portion to a solution of 5- tert-butyl-7-(3,5-dimethylphenyl)-2-isopropyl-6-methoxy-1H-indene (11.42 g, 32.77 mmol) in 200 ml of ether cooled to –50 °C. This mixture was stirred overnight at room temperature, then the resulting yellow suspension was cooled to –30 °C, and 10 ml of THF followed by 250 mg of CuCN were added. The obtained mixture was stirred for 0.5 h at –25 °C, then a solution of [6-tert-butyl-4-(3,5-dimethylphenyl)-2-methyl-5-methoxy-1H-inden-1- yl](chloro)dimethylsilane (13.53 g, 32.76 mmol) in 200 ml of Et2O was added in one portion. This mixture was stirred overnight at room temperature, then filtered through a pad of silica gel 60 (40-63 µm), which was additionally washed by 2 ^50 ml of Et2O. The combined organic elute was evaporated to dryness, and the residue was dried in vacuum at elevated temperature to give 23.83 g (ca.100% of ca. 90% purity) of the title product (ca.80:20 mixture of the stereoisomers) as a yellowish glassy solid which was used without further purification. 1H NMR (CDCl3): δ 7.33 and 7.29 (2s, sum 2H), 7.12 (s, 4H), 6.99 (2s, sum 2H), 6.47 and 6.42 (2s, sum 2H), 3.64 and 3.55 (2s, sum 2H), 3.27, 3.26, 3.25 and 3.24 (4s, sum 6H), 2.67-2.44 (m, 1H), 2.39 and 2.38 (2s, sum 12H), 2.16 and 2.05 (2s, sum 3H), 1.44 (3s) and 1.43 (s) {sum 18H}, 1.16 ( d, J = 6.7 Hz, 3H), 1.03 (d, J = 6.8 Hz) and 1.01 (d, J = 6.8 Hz) {sum 3H}, -0.10, -0.16, -0.23 and -0.24 (4s, sum 6H).
nBuLi in hexanes (2.5 M,13.1 ml, 32.75 mmol) was added in one portion to a solution of 5- tert-butyl-7-(3,5-dimethylphenyl)-2-isopropyl-6-methoxy-1H-indene (11.42 g, 32.77 mmol) in 200 ml of ether cooled to –50 °C. This mixture was stirred overnight at room temperature, then the resulting yellow suspension was cooled to –30 °C, and 10 ml of THF followed by 250 mg of CuCN were added. The obtained mixture was stirred for 0.5 h at –25 °C, then a solution of [6-tert-butyl-4-(3,5-dimethylphenyl)-2-methyl-5-methoxy-1H-inden-1- yl](chloro)dimethylsilane (13.53 g, 32.76 mmol) in 200 ml of Et2O was added in one portion. This mixture was stirred overnight at room temperature, then filtered through a pad of silica gel 60 (40-63 µm), which was additionally washed by 2 ^50 ml of Et2O. The combined organic elute was evaporated to dryness, and the residue was dried in vacuum at elevated temperature to give 23.83 g (ca.100% of ca. 90% purity) of the title product (ca.80:20 mixture of the stereoisomers) as a yellowish glassy solid which was used without further purification. 1H NMR (CDCl3): δ 7.33 and 7.29 (2s, sum 2H), 7.12 (s, 4H), 6.99 (2s, sum 2H), 6.47 and 6.42 (2s, sum 2H), 3.64 and 3.55 (2s, sum 2H), 3.27, 3.26, 3.25 and 3.24 (4s, sum 6H), 2.67-2.44 (m, 1H), 2.39 and 2.38 (2s, sum 12H), 2.16 and 2.05 (2s, sum 3H), 1.44 (3s) and 1.43 (s) {sum 18H}, 1.16 ( d, J = 6.7 Hz, 3H), 1.03 (d, J = 6.8 Hz) and 1.01 (d, J = 6.8 Hz) {sum 3H}, -0.10, -0.16, -0.23 and -0.24 (4s, sum 6H).
 anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1- yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride
anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1- yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride
 nBuLi in hexanes (2.5 M, 8.0 ml, 20.0 mmol) was added in one portion at room temperature to a yellowish solution of [6-tert-butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-isopropyl-1H- inden-1-yl][6-tert-butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-methyl-1H-inden-1- yl]dimethylsilane (7.25 g, ca. 10.0 mmol) in 50 ml of nBu2O. This mixture was stirred overnight at room temperature, then the resulting red solution was cooled to 0 °C in an ice- bath, and ZrCl4 (2.33 g, 9.99 mmol) was added. The reaction mixture was stirred for 24 h at room temperature to give a red solution with precipitate of LiCl. This mixture was evaporated to dryness, and the residue was treated with 50 ml of warm toluene. The obtained suspension was filtered through glass frit (G4), the filter cake was washed with 2 ^20 ml of warm toluene. On the evidence of NMR spectroscopy, the resulting filtrate included a ca. 52/48 mixture of anti- and syn-zirconocene dichlorides. The filtrate was evaporated to the state of oil, and this oil was dissolved in 30 ml of n-pentane. The orange precipitate fallen from this solution shortly after dissolution was filtered off, washed with 5 ml of n-pentane, and dried in vacuum. This procedure gave 2.0 g of syn- dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1- yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride contaminated with 2% of anti-isomer. After standing overnight at room temperature, 300 mg of a mixture of syn-/anti- = 1/1 fell out of the mother liquor. The mother liquor was evaporated to dryness, and the residue was dissolved in 25 ml of n-pentane. Orange powder precipitated from this solution for 2 days at –25 °C was collected and then dried under vacuum. This procedure gave 1.98 g of anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-
dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl][2-methyl-4-(3,5-dimethylphenyl)-5- methoxy-6-tert-butylinden-1-yl]zirconium dichloride containing 1 equiv. of n-pentane (or 0.15 g of n-pentane in 1.98 g of the product), so the adjusted net weight of the isolated anti-complex was 1.83 g. Thus, the total yield of syn- and anti- dimethylsilanediyl[2-iso- propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl][2-methyl-4-(3,5- dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichlorides isolated in this synthesis was 4.13 g (46.7%). Syn-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden- 1-yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride. 1H NMR (CDCl3): δ 7.74-7.34 (very br.s, 2H), 7.41 (s, 2H), 7.33-6.93 (very br.s, 2H), 6.99 (s, 1H), 6.97 (s, 1H), 6.63 (s, 1H), 6.58 (s, 1H), 3.30 (m, 1H), 3.28 (s, 3H), 3.25 (s, 3H), 2.38 (s, 6H), 2.36 (s, 6H), 2.31 (s, 3H), 1.41 (s, 3H), 1.37 (s, 18H), 1.32 (d, J = 6.7 Hz, 3H), 1.20 (s, 3H), 1.09 (d, J = 6.7 Hz, 3H). Anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden- 1-yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride x 1.0 npentane. Anal. calc. for C50H62Cl2O2SiZr x C5H12: C, 69.00; H, 7.79. Found: C, 69.27; H, 7.95. 1H NMR (CDCl3): δ 7.70-6.80 (2 very br.s, sum 4H), 7.52 (s, 1H), 7.45 (s, 1H), 6.98 (s, 1H), 6.96 (s, 1H), 6.61 (s, 1H), 6.57 (s, 1H), 3.44 (s, 3H), 3.42 (s, 3H), 3.19 (sept, J = 6.7 Hz, 1H), 2.36 (s, 6H), 2.35 (s, 6H), 2.15 (s, 3H), 1.38 (s, 18H), 1.29 (s, 6H), 1.09 (d, J = 6.7 Hz, 3H), 1.03 (d, J = 6.7 Hz, 3H).
nBuLi in hexanes (2.5 M, 8.0 ml, 20.0 mmol) was added in one portion at room temperature to a yellowish solution of [6-tert-butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-isopropyl-1H- inden-1-yl][6-tert-butyl-4-(3,5-dimethylphenyl)-5-methoxy-2-methyl-1H-inden-1- yl]dimethylsilane (7.25 g, ca. 10.0 mmol) in 50 ml of nBu2O. This mixture was stirred overnight at room temperature, then the resulting red solution was cooled to 0 °C in an ice- bath, and ZrCl4 (2.33 g, 9.99 mmol) was added. The reaction mixture was stirred for 24 h at room temperature to give a red solution with precipitate of LiCl. This mixture was evaporated to dryness, and the residue was treated with 50 ml of warm toluene. The obtained suspension was filtered through glass frit (G4), the filter cake was washed with 2 ^20 ml of warm toluene. On the evidence of NMR spectroscopy, the resulting filtrate included a ca. 52/48 mixture of anti- and syn-zirconocene dichlorides. The filtrate was evaporated to the state of oil, and this oil was dissolved in 30 ml of n-pentane. The orange precipitate fallen from this solution shortly after dissolution was filtered off, washed with 5 ml of n-pentane, and dried in vacuum. This procedure gave 2.0 g of syn- dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1- yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride contaminated with 2% of anti-isomer. After standing overnight at room temperature, 300 mg of a mixture of syn-/anti- = 1/1 fell out of the mother liquor. The mother liquor was evaporated to dryness, and the residue was dissolved in 25 ml of n-pentane. Orange powder precipitated from this solution for 2 days at –25 °C was collected and then dried under vacuum. This procedure gave 1.98 g of anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-
dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl][2-methyl-4-(3,5-dimethylphenyl)-5- methoxy-6-tert-butylinden-1-yl]zirconium dichloride containing 1 equiv. of n-pentane (or 0.15 g of n-pentane in 1.98 g of the product), so the adjusted net weight of the isolated anti-complex was 1.83 g. Thus, the total yield of syn- and anti- dimethylsilanediyl[2-iso- propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl][2-methyl-4-(3,5- dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichlorides isolated in this synthesis was 4.13 g (46.7%). Syn-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden- 1-yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride. 1H NMR (CDCl3): δ 7.74-7.34 (very br.s, 2H), 7.41 (s, 2H), 7.33-6.93 (very br.s, 2H), 6.99 (s, 1H), 6.97 (s, 1H), 6.63 (s, 1H), 6.58 (s, 1H), 3.30 (m, 1H), 3.28 (s, 3H), 3.25 (s, 3H), 2.38 (s, 6H), 2.36 (s, 6H), 2.31 (s, 3H), 1.41 (s, 3H), 1.37 (s, 18H), 1.32 (d, J = 6.7 Hz, 3H), 1.20 (s, 3H), 1.09 (d, J = 6.7 Hz, 3H). Anti-dimethylsilanediyl[2-iso-propyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden- 1-yl][2-methyl-4-(3,5-dimethylphenyl)-5-methoxy-6-tert-butylinden-1-yl]zirconium dichloride x 1.0 npentane. Anal. calc. for C50H62Cl2O2SiZr x C5H12: C, 69.00; H, 7.79. Found: C, 69.27; H, 7.95. 1H NMR (CDCl3): δ 7.70-6.80 (2 very br.s, sum 4H), 7.52 (s, 1H), 7.45 (s, 1H), 6.98 (s, 1H), 6.96 (s, 1H), 6.61 (s, 1H), 6.57 (s, 1H), 3.44 (s, 3H), 3.42 (s, 3H), 3.19 (sept, J = 6.7 Hz, 1H), 2.36 (s, 6H), 2.35 (s, 6H), 2.15 (s, 3H), 1.38 (s, 18H), 1.29 (s, 6H), 1.09 (d, J = 6.7 Hz, 3H), 1.03 (d, J = 6.7 Hz, 3H).
 wherein Mt is Zr or Hf, preferably Zr; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C3-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; n are each independently an integer from 1 to 5; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl group, or a –OR31, -SR31 or -NR312 group in which R31 is C1-C10- hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; L is O or S; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or - OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or
adjacent LR51 and R6 and/or LR51’ and R6’ form together a -O[C(R62)2]mO- group, with each R62 being independently, same or different from each other, H or linear or branched C1-C6-alkyl, with m being 1 to 3. 2. A metallocene complex of formula (I) as claimed in claim 1 having formula (I-a)
wherein Mt is Zr or Hf, preferably Zr; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C3-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; n are each independently an integer from 1 to 5; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl group, or a –OR31, -SR31 or -NR312 group in which R31 is C1-C10- hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; L is O or S; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or - OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or
adjacent LR51 and R6 and/or LR51’ and R6’ form together a -O[C(R62)2]mO- group, with each R62 being independently, same or different from each other, H or linear or branched C1-C6-alkyl, with m being 1 to 3. 2. A metallocene complex of formula (I) as claimed in claim 1 having formula (I-a)
 wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl group, or a –OR31, -SR31 or -NR312 group in which R31 is C1-C10- hydrocarbyl, whereby at least one R3 and at least one R4 is not H; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or - OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or
adjacent OR51 and R6 and/or OR51’ and R6’ form together a -O [C(R62)2]m O- group, with each R62 being independently, same or different from each other, H or linear or branched C1-C6-alkyl, with m being 1 to 3. 3. A metallocene complex of formula (I) as claimed in claim 1 or 2 having formula (I-b)
wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, H, C1-C10- hydrocarbyl group, or a –OR31, -SR31 or -NR312 group in which R31 is C1-C10- hydrocarbyl, whereby at least one R3 and at least one R4 is not H; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or - OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or
adjacent OR51 and R6 and/or OR51’ and R6’ form together a -O [C(R62)2]m O- group, with each R62 being independently, same or different from each other, H or linear or branched C1-C6-alkyl, with m being 1 to 3. 3. A metallocene complex of formula (I) as claimed in claim 1 or 2 having formula (I-b)
 (I-b) wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R3 and R4 are each independently H, C1-C10-hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or - OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR51 and R6 and/or OR51’ and R6’ form together a -O[C(R62)2]mO- group, with R62 being each independently, same or different from each other, independently H or linear or branched C1-C6-alkyl, with m being 1 to 3. 4. A metallocene complex of formula (I) as claimed in any one of claims 1 to 3 having formula (I-c)
(I-b) wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R3 and R4 are each independently H, C1-C10-hydrocarbyl, -OR31, -SR31, or –N(R31)2, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3 or - OR61, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl; or adjacent OR51 and R6 and/or OR51’ and R6’ form together a -O[C(R62)2]mO- group, with R62 being each independently, same or different from each other, independently H or linear or branched C1-C6-alkyl, with m being 1 to 3. 4. A metallocene complex of formula (I) as claimed in any one of claims 1 to 3 having formula (I-c)
 (I-c) wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, C1-C10- hydrocarbyl group, or a –OR31, -SR31 or –N(R31)2 group in which R31 is C1-C10- hydrocarbyl; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl. 5. A metallocene complex of formula (I) as claimed in any one of claims 1 to 4 having formula (I-d)
(I-c) wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently, same or different from each other, C1-C10- hydrocarbyl group, or a –OR31, -SR31 or –N(R31)2 group in which R31 is C1-C10- hydrocarbyl; R51 and R51’ are each independently, same of different from each other, C1-C10- hydrocarbyl; and R6 and R6’ are each independently, same or different from each other, C(R61)3, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl. 5. A metallocene complex of formula (I) as claimed in any one of claims 1 to 4 having formula (I-d)
 wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently C1-C10-hydrocarbyl group, or a –OR31, -SR31 or - NR31 2 group, with R31 being C1-C10-hydrocarbyl. 6. A metallocene complex of formula (I) as claimed in any one of claims 1 to 4 having formula (I-e)
wherein Mt is Zr or Hf; X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being each independently, same or different from each other, C1-C10-hydrocarbyl; R3 and R4 are each independently C1-C10-hydrocarbyl group, or a –OR31, -SR31 or - NR31 2 group, with R31 being C1-C10-hydrocarbyl. 6. A metallocene complex of formula (I) as claimed in any one of claims 1 to 4 having formula (I-e)
 wherein
X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being C1-C10- hydrocarbyl; R3 and R4 are each independently H, C1-C10-hydrocarbyl group, or a –OR31, -SR31 or -NR31 2 group, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen. 7. A metallocene complex as claimed in any one of claims 1 to 6, wherein one or two R3 on the phenyl group are not H and the R3 are the same, and two R4 on the phenyl group are not H and these two R4 are the same. 8. A polymerization catalyst, comprising, preferably consisting of (i) a metallocene complex of formula (I) as claimed in any one of claim 1 to 6; (ii) a cocatalyst comprising a group 13 element; and (iii) optionally a support. 9. The polymerization catalyst according to claim 7, wherein cocatalyst (ii) is an alumoxane cocatalyst, preferably in the absence of any further cocatalysts. 10. The catalyst system as claimed in claim 7 or 8, supported on silica. 11. A process for the polymerization of propylene, comprising polymerizing propylene and optionally at least one of comonomer selected from ethylene or C4-10 alpha olefin comonomers s in the presence of the metallocene catalyst claimed in any one of claim 8 to 10. 12. A process for the polymerization of propylene, comprising polymerizing propylene and optionally at least one of comonomer selected from ethylene or C4-C10-alpha olefin comonomers s in the presence of the metallocene catalyst claimed in any one of claim 8 to 10. 13. A process for the preparation of a heterophasic polypropylene copolymer comprising
(I) polymerizing propylene in bulk in the presence of a polymerization catalyst as claimed in claim 8 to 10 to form a polypropylene homopolymer matrix; (II) in the presence of said matrix and said polymerization catalyst and in the gas phase, polymerizing propylene and ethylene to form a heterophasic polypropylene copolymer comprising a homopolymer matrix and an ethylene propylene rubber. 14. A process for the preparation of a heterophasic polypropylene copolymer comprising (I) polymerizing propylene in bulk in the presence of polymerization catalyst as claimed in claim 8 to 10 to form a polypropylene homopolymer; (II) in the presence of said homopolymer and said polymerization catalyst and in the gas phase, polymerizing propylene to form a polypropylene homopolymer matrix; (III) in the presence said matrix and said polymerization catalyst and in the gas phase, polymerizing propylene and ethylene to form a heterophasic polypropylene copolymer comprising a homopolymer matrix and an ethylene propylene rubber (EPR). 15. An indene of formula (II)
wherein
X is a sigma ligand R1 are each independently, same or different from each other, C1-C20-hydrocarbyl, optionally containing up to two heteroatoms of Group 14-16 of the Periodic Table, or form together with the Si atom they are attached to a C4-C8-ring; R2 is linear C1-C20-hydrocarbyl; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being C1-C10- hydrocarbyl; R3 and R4 are each independently H, C1-C10-hydrocarbyl group, or a –OR31, -SR31 or -NR31 2 group, with R31 being C1-C10-hydrocarbyl, whereby at least one R3 and at least one R4 is not hydrogen. 7. A metallocene complex as claimed in any one of claims 1 to 6, wherein one or two R3 on the phenyl group are not H and the R3 are the same, and two R4 on the phenyl group are not H and these two R4 are the same. 8. A polymerization catalyst, comprising, preferably consisting of (i) a metallocene complex of formula (I) as claimed in any one of claim 1 to 6; (ii) a cocatalyst comprising a group 13 element; and (iii) optionally a support. 9. The polymerization catalyst according to claim 7, wherein cocatalyst (ii) is an alumoxane cocatalyst, preferably in the absence of any further cocatalysts. 10. The catalyst system as claimed in claim 7 or 8, supported on silica. 11. A process for the polymerization of propylene, comprising polymerizing propylene and optionally at least one of comonomer selected from ethylene or C4-10 alpha olefin comonomers s in the presence of the metallocene catalyst claimed in any one of claim 8 to 10. 12. A process for the polymerization of propylene, comprising polymerizing propylene and optionally at least one of comonomer selected from ethylene or C4-C10-alpha olefin comonomers s in the presence of the metallocene catalyst claimed in any one of claim 8 to 10. 13. A process for the preparation of a heterophasic polypropylene copolymer comprising
(I) polymerizing propylene in bulk in the presence of a polymerization catalyst as claimed in claim 8 to 10 to form a polypropylene homopolymer matrix; (II) in the presence of said matrix and said polymerization catalyst and in the gas phase, polymerizing propylene and ethylene to form a heterophasic polypropylene copolymer comprising a homopolymer matrix and an ethylene propylene rubber. 14. A process for the preparation of a heterophasic polypropylene copolymer comprising (I) polymerizing propylene in bulk in the presence of polymerization catalyst as claimed in claim 8 to 10 to form a polypropylene homopolymer; (II) in the presence of said homopolymer and said polymerization catalyst and in the gas phase, polymerizing propylene to form a polypropylene homopolymer matrix; (III) in the presence said matrix and said polymerization catalyst and in the gas phase, polymerizing propylene and ethylene to form a heterophasic polypropylene copolymer comprising a homopolymer matrix and an ethylene propylene rubber (EPR). 15. An indene of formula (II)
 wherein the dotted lines represent a double bond present in between carbons 1 and 2 or 2 and 3 of the indenyl ring; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being C1-C10- hydrocarbyl; R4 are each independently H, C1-C10-hydrocarbyl group, or a –OR31, -SR31 or -NR31 2 group in which R31 is C1-C10-hydrocarbyl, whereby at least one R4 is not H; R51’ is C1-C10-hydrocarbyl; and R6’ is C(R61)3, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl.
wherein the dotted lines represent a double bond present in between carbons 1 and 2 or 2 and 3 of the indenyl ring; R2’ is alpha-branched C1-C10-hydrocarbyl or SiH(R21’)2, with R21’ being C1-C10- hydrocarbyl; R4 are each independently H, C1-C10-hydrocarbyl group, or a –OR31, -SR31 or -NR31 2 group in which R31 is C1-C10-hydrocarbyl, whereby at least one R4 is not H; R51’ is C1-C10-hydrocarbyl; and R6’ is C(R61)3, with R61 being each independently, same of different from each other, linear or branched C1-C6-alkyl.